
11.1 Principles of Biotechnology
11.2 Tools of Recombinant DNA
Technology
11.3 Processes of Recombinant
DNA Technology
Biotechnology deals with techniques of using live
organisms or enzymes from organisms to produce products
and processes useful to humans. In this sense, making
curd, bread or wine, which are all microbe-mediated
processes, could also be thought as a form of
biotechnology. However, it is used in a restricted sense
today, to refer to such of those processes which use
genetically modified organisms to achieve the same on a
larger scale. Further, many other processes/techniques are
also included under biotechnology. For example, in vitro
fertilisation leading to a ‘test-tube’ baby, synthesising a
gene and using it, developing a DNA vaccine or correcting
a defective gene, are all part of biotechnology.
The European Federation of Biotechnology (EFB) has
given a definition of biotechnology that encompasses both
traditional view and modern molecular biotechnology.
The definition given by EFB is as follows:
‘The integration of natural science and organisms,
cells, parts thereof, and molecular analogues for products
and services’.
11.1 PRINCIPLES OF BIOTECHNOLOGY
Among many, the two core techniques that enabled birth
of modern biotechnology are :
(i) Genetic engineering : Techniques to alter the
chemistry of genetic material (DNA and RNA),
to introduce these into host organisms and thus change the
phenotype of the host organism.
(ii) Bioprocess engineering: Maintenance of sterile (microbial
contamination-free) ambience in chemical engineering processes
to enable growth of only the desired microbe/eukaryotic cell in
large quantities for the manufacture of biotechnological products
like antibiotics, vaccines, enzymes, etc.
Let us now understand the conceptual development of the principles
of genetic engineering.
You probably appreciate the advantages of sexual reproduction over
asexual reproduction. The former provides opportunities for variations
and formulation of unique combinations of genetic setup, some of which
may be beneficial to the organism as well as the population. Asexual
reproduction preserves the genetic information, while sexual reproduction
permits variation. Traditional hybridisation procedures used in plant and
animal breeding, very often lead to inclusion and multiplication of
undesirable genes along with the desired genes. The techniques of genetic
engineering which include creation of recombinant DNA, use of
gene cloning and gene transfer, overcome this limitation and allows us
to isolate and introduce only one or a set of desirable genes without
introducing undesirable genes into the target organism.
Do you know the likely fate of a piece of DNA, which is somehow
transferred into an alien organism? Most likely, this piece of DNA would
not be able to multiply itself in the progeny cells of the organism. But,
when it gets integrated into the genome of the recipient, it may multiply
and be inherited along with the host DNA. This is because the alien piece
of DNA has become part of a chromosome, which has the ability to
replicate. In a chromosome there is a specific DNA sequence called the
origin of replication, which is responsible for initiating replication.
Therefore, for the multiplication of any alien piece of DNA in an organism
it needs to be a part of a chromosome(s) which has a specific sequence
known as ‘origin of replication’. Thus, an alien DNA is linked with the
origin of replication, so that, this alien piece of DNA can replicate and
multiply itself in the host organism. This can also be called as cloning or
making multiple identical copies of any template DNA.
Let us now focus on the first instance of the construction of an artificial
recombinant DNA molecule. The construction of the first recombinant
DNA emerged from the possibility of linking a gene encoding antibiotic
resistance with a native plasmid (autonomously replicating circular
extra-chromosomal DNA) of Salmonella typhimurium. Stanley Cohen and
Herbert Boyer accomplished this in 1972 by isolating the antibiotic
resistance gene by cutting out a piece of DNA from a plasmid which was
responsible for conferring antibiotic resistance. The cutting of DNA at
specific locations became possible with the discovery of the so-called
‘molecular scissors’– restriction enzymes. The cut piece of DNA was
then linked with the plasmid DNA. These plasmid DNA act as vectors to
transfer the piece of DNA attached to it. You probably know that mosquito
acts as an insect vector to transfer the malarial parasite into human body.
In the same way, a plasmid can be used as vector to deliver an alien piece
of DNA into the host organism. The linking of antibiotic resistance gene
with the plasmid vector became possible with the enzyme DNA ligase,
which acts on cut DNA molecules and joins their ends. This makes a new
combination of circular autonomously replicating DNA created in vitro
and is known as recombinant DNA. When this DNA is transferred into
Escherichia coli, a bacterium closely related to Salmonella, it could
replicate using the new host’s DNA polymerase enzyme and make multiple
copies. The ability to multiply copies of antibiotic resistance gene in
E. coli was called cloning of antibiotic resistance gene in E. coli.
You can hence infer that there are three basic steps in genetically
modifying an organism —
(i) identification of DNA with desirable genes;
(ii) introduction of the identified DNA into the host;
(iii) maintenance of introduced DNA in the host and transfer of the DNA
to its progeny.
11.2 TOOLS OF RECOMBINANT DNA TECHNOLOGY
Now we know from the foregoing discussion that genetic engineering or
recombinant DNA technology can be accomplished only if we have the
key tools, i.e., restriction enzymes, polymerase enzymes, ligases, vectors
and the host organism. Let us try to understand some of these in detail.
11.2.1 Restriction Enzymes
In the year 1963, the two enzymes responsible for restricting the growth
of bacteriophage in Escherichia coli were isolated. One of these added
methyl groups to DNA, while the other cut DNA. The later was called
restriction endonuclease.
The first restriction endonuclease–Hind II, whose functioning
depended on a specific DNA nucleotide sequence was isolated and
characterised five years later. It was found that Hind II always cut DNA
molecules at a particular point by recognising a specific sequence of
six base pairs. This specific base sequence is known as the
recognition sequence for Hind II. Besides Hind II, today we know more
than 900 restriction enzymes that have been isolated from over 230 strains
of bacteria each of which recognise different recognition sequences.
The convention for naming these enzymes is the first letter of the name
comes from the genus and the second two letters come from the species of
the prokaryotic cell from which they were isolated, e.g., EcoRI comes from
Escherichia coli RY 13. In EcoRI, the letter ‘R’ is derived from the name of
strain. Roman numbers following the names indicate the order in which
the enzymes were isolated from that strain of bacteria.
Restriction enzymes belong to a larger class of enzymes called
nucleases. These are of two kinds; exonucleases and endonucleases.
Exonucleases remove nucleotides from the ends of the DNA whereas,
endonucleases make cuts at specific positions within the DNA.
Each restriction endonuclease functions by ‘inspecting’ the length of
a DNA sequence. Once it finds its specific recognition sequence, it
will bind to the DNA and cut each of the two strands of the double
helix at specific points in their sugar -phosphate backbones
(Figure 11.1). Each restriction endonuclease recognises a specific
palindromic nucleotide sequences in the DNA.
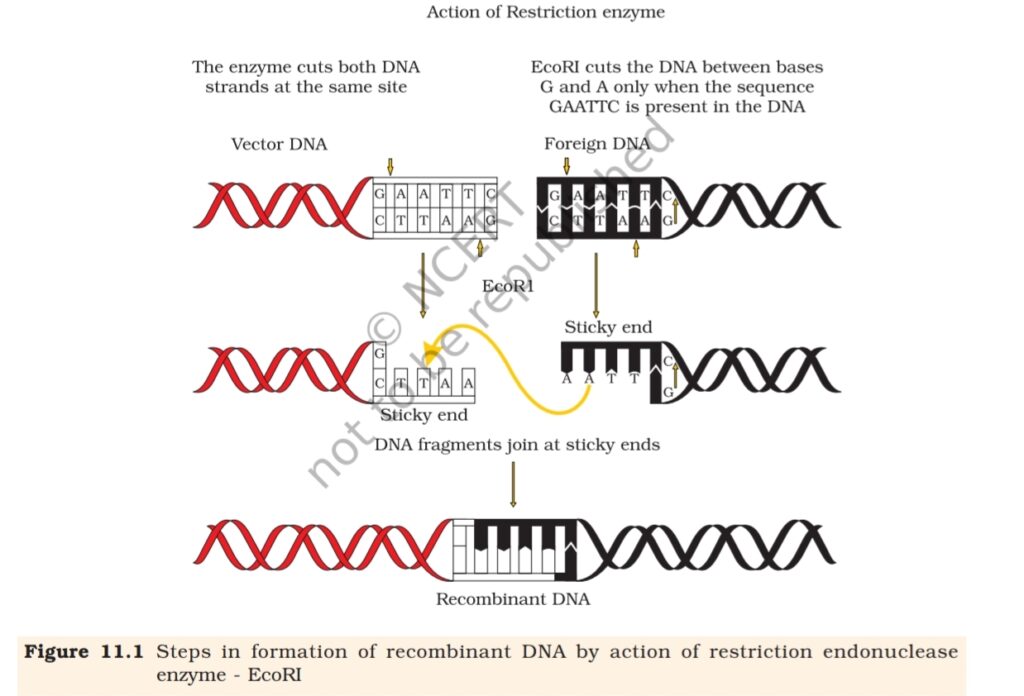
Do you know what palindromes are? These are groups of letters
that form the same words when read both forward and backward,
e.g., “MALAYALAM”. As against a word-palindrome where the same
word is read in both directions, the palindrome in DNA is a sequence
of base pairs that reads same on the two strands when orientation of
reading is kept the same. For example, the following sequences reads
the same on the two strands in 5′ à 3′ direction. This is also true if
read in the 3′ à 5′ direction.
5′ —— GAATTC —— 3′
3′ —— CTTAAG —— 5′
Restriction enzymes cut the strand of DNA a little away from the centre
of the palindrome sites, but between the same two bases on the opposite
strands. This leaves single stranded portions at the ends. There are
overhanging stretches called sticky ends on each strand (Figure 11.1).
These are named so because they form hydrogen bonds with their
complementary cut counterparts. This stickiness of the ends facilitates
the action of the enzyme DNA ligase.
Restriction endonucleases are used in genetic engineering to form
‘recombinant’ molecules of DNA, which are composed of DNA from
different sources/genomes.
When cut by the same restriction enzyme, the resultant DNA fragments
have the same kind of ‘sticky-ends’ and, these can be joined together
(end-to-end) using DNA ligases (Figure 11.2).
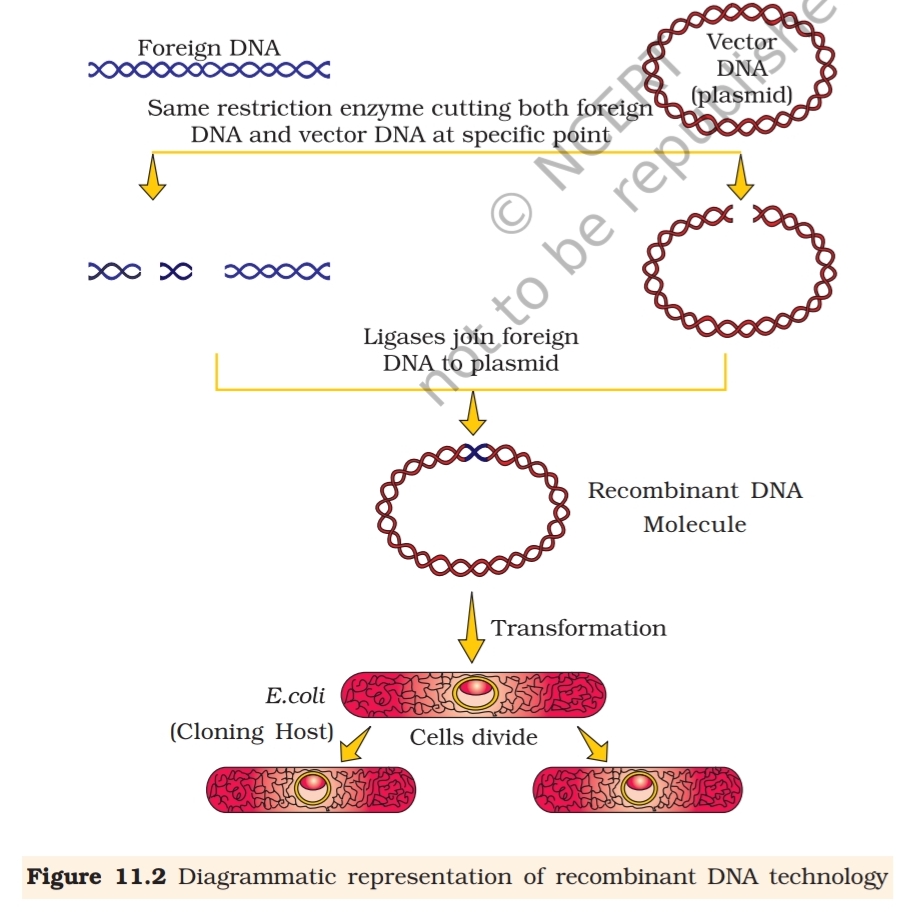
You may have realised that normally, unless one cuts the vector and
the source DNA with the same restriction enzyme, the recombinant vector
molecule cannot be created.
Separation and isolation of DNA fragments : The cutting of DNA by
restriction endonucleases results in the fragments of DNA. These fragments
can be separated by a technique known as gel electrophoresis. Since
DNA fragments are negatively charged molecules they can be separated
by forcing them to move towards the anode under an electric field through
a medium/matrix. Nowadays the most commonly used matrix is agarose
which is a natural polymer extracted from sea weeds. The DNA fragments
separate (resolve) according to their size through sieving effect provided
by the agarose gel. Hence, the smaller the fragment size, the farther it
moves. Look at the Figure 11.3 and guess at which end of the gel the
sample was loaded.
You may have realised that normally, unless one cuts the vector and
the source DNA with the same restriction enzyme, the recombinant vector
molecule cannot be created.
Separation and isolation of DNA fragments : The cutting of DNA by
restriction endonucleases results in the fragments of DNA. These fragments
can be separated by a technique known as gel electrophoresis. Since
DNA fragments are negatively charged molecules they can be separated
by forcing them to move towards the anode under an electric field through
a medium/matrix. Nowadays the most commonly used matrix is agarose
which is a natural polymer extracted from sea weeds. The DNA fragments
separate (resolve) according to their size through sieving effect provided
by the agarose gel. Hence, the smaller the fragment size, the farther it
moves. Look at the Figure 11.3 and guess at which end of the gel the
sample was loaded.
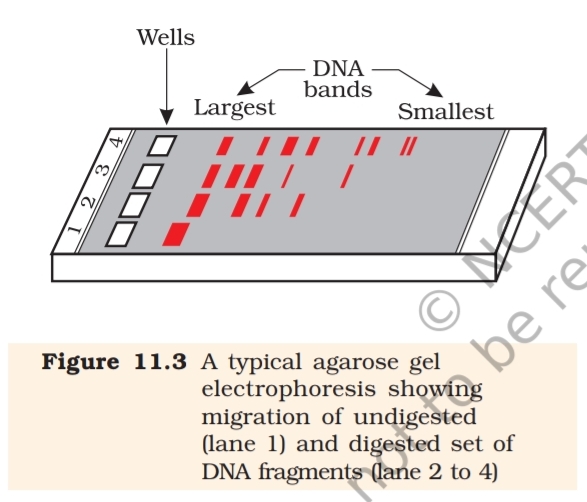
The separated DNA fragments can be
visualised only after staining the DNA
with a compound known as ethidium
bromide followed by exposure to UV
radiation (you cannot see pure DNA
fragments in the visible light and
without staining). You can see bright
orange coloured bands of DNA in a
ethidium bromide stained gel
exposed to UV light (Figure 11.3). The
separated bands of DNA are cut out
from the agarose gel and extracted
from the gel piece. This step is known
as elution. The DNA fragments
purified in this way are used in
constructing recombinant DNA by
joining them with cloning vectors.
11.2.2 Cloning Vectors
You know that plasmids and bacteriophages have the ability to replicate
within bacterial cells independent of the control of chromosomal DNA.
Bacteriophages because of their high number per cell, have very high
copy numbers of their genome within the bacterial cells. Some plasmids
may have only one or two copies per cell whereas others may have
15-100 copies per cell. Their numbers can go even higher. If we are able
to link an alien piece of DNA with bacteriophage or plasmid DNA, we can
multiply its numbers equal to the copy number of the plasmid or
bacteriophage. Vectors used at present, are engineered in such a way
that they help easy linking of foreign DNA and selection of recombinants
from non-recombinants.
The following are the features that are required to facilitate cloning
into a vector.

(i) Origin of replication (ori) : This is a sequence from where
replication starts and any piece of DNA when linked to this sequence
can be made to replicate within the host cells. This sequence is also
responsible for controlling the copy number of the linked DNA. So,
if one wants to recover many copies of the target DNA it should be
cloned in a vector whose origin support high copy number.
(ii) Selectable marker : In addition to ‘ori’, the vector requires a
selectable marker, which helps in identifying and eliminating non-
transformants and selectively permitting the growth of the
transformants. Transformation is a procedure through which a
piece of DNA is introduced in a host bacterium (you will study the
process in subsequent section). Normally, the genes encoding
resistance to antibiotics such as ampicillin, chloramphenicol,
tetracycline or kanamycin, etc., are considered useful selectable
markers for E. coli. The normal E. coli cells do not carry resistance
against any of these antibiotics.
(iii) Cloning sites: In order to link the
alien DNA, the vector needs to have
very few, preferably single,
recognition sites for the commonly
used restriction enzymes. Presence of
more than one recognition sites within
the vector will generate several
fragments, which will complicate the
gene cloning (Figure 11.4). The
ligation of alien DNA is carried out at
a restriction site present in one of the
two antibiotic resistance genes. For
example, you can ligate a foreign DNA
at the BamH I site of tetracycline
resistance gene in the vector pBR322.
The recombinant plasmids will lose
tetracycline resistance due to insertion
of foreign DNA but can still be selected
out from non-recombinant ones by
plating the transformants on
tetracycline containing medium. The transformants growing on
ampicillin containing medium are then transferred on a medium
containing tetracycline. The recombinants will grow in ampicillin
containing medium but not on that containing tetracycline. But,
non- recombinants will grow on the medium containing both the
antibiotics. In this case, one antibiotic resistance gene helps in
selecting the transformants, whereas the other antibiotic resistance
gene gets ‘inactivated due to insertion’ of alien DNA, and helps in
selection of recombinants.
Selection of recombinants due to inactivation of antibiotics is a
cumbersome procedure because it requires simultaneous plating
on two plates having different antibiotics. Therefore, alternative
selectable markers have been developed which differentiate
recombinants from non-recombinants on the basis of their ability
to produce colour in the presence of a chromogenic substrate. In
this, a recombinant DNA is inserted within the coding sequence of
an enzyme, β-galactosidase. This results into inactivation of the
gene for synthesis of this enzyme, which is referred to as insertional
inactivation. The presence of a chromogenic substrate gives blue
coloured colonies if the plasmid in the bacteria does not have an
insert. Presence of insert results into insertional inactivation of the
β-galactosidase gene and the colonies do not produce any colour,
these are identified as recombinant colonies.
(iv) Vectors for cloning genes in plants and animals : You may be
surprised to know that we have learnt the lesson of transferring genes
into plants and animals from bacteria and viruses which have known
this for ages – how to deliver genes to transform eukaryotic cells and
force them to do what the bacteria or viruses want. For example,
Agrobacterium tumifaciens, a pathogen of several dicot plants is able
to deliver a piece of DNA known as ‘T-DNA’ to transform normal
plant cells into a tumor and direct these tumor cells to produce the
chemicals required by the pathogen. Similarly, retroviruses in animals
have the ability to transform normal cells into cancerous cells. A
better understanding of the art of delivering genes by pathogens in
their eukaryotic hosts has generated knowledge to transform these
tools of pathogens into useful vectors for delivering genes of interest
to humans. The tumor inducing (Ti) plasmid of Agrobacterium
tumifaciens has now been modified into a cloning vector which is no
more pathogenic to the plants but is still able to use the mechanisms
to deliver genes of our interest into a variety of plants. Similarly,
retroviruses have also been disarmed and are now used to deliver
desirable genes into animal cells. So, once a gene or a DNA fragment
has been ligated into a suitable vector it is transferred into a bacterial,
plant or animal host (where it multiplies).
11.2.3 Competent Host (For Transformation with
Recombinant DNA)
Since DNA is a hydrophilic molecule, it cannot pass through cell
membranes. Why? In order to force bacteria to take up the plasmid, the
bacterial cells must first be made ‘competent’ to take up DNA. This is
done by treating them with a specific concentration of a divalent cation,
such as calcium, which increases the efficiency with which DNA enters
the bacterium through pores in its cell wall. Recombinant DNA can then
be forced into such cells by incubating the cells with recombinant DNA
on ice, followed by placing them briefly at 420C (heat shock), and then
putting them back on ice. This enables the bacteria to take up the
recombinant DNA.
This is not the only way to introduce alien DNA into host cells. In a
method known as micro-injection, recombinant DNA is directly injected
into the nucleus of an animal cell. In another method, suitable for plants,
cells are bombarded with high velocity micro-particles of gold or tungsten
coated with DNA in a method known as biolistics or gene gun. And the
last method uses ‘disarmed pathogen’ vectors, which when allowed to
infect the cell, transfer the recombinant DNA into the host.
Now that we have learnt about the tools for constructing recombinant
DNA, let us discuss the processes facilitating recombinant DNA technology.
11.3 PROCESSES OF RECOMBINANT DNA TECHNOLOGY
Recombinant DNA technology involves several steps in specific
sequence such as isolation of DNA, fragmentation of DNA by
restriction endonucleases, isolation of a desired DNA fragment,
ligation of the DNA fragment into a vector, transferring the
recombinant DNA into the host, culturing the host cells in a
medium at large scale and extraction of the desired product.
Let us examine each of these steps in some details.
11.3.1 Isolation of the Genetic Material (DNA)
Recall that nucleic acid is the genetic material of all organisms
without exception. In majority of organisms this is
deoxyribonucleic acid or DNA. In order to cut the DNA with
restriction enzymes, it needs to be in pure form, free from other
macro-molecules. Since the DNA is enclosed within the
membranes, we have to break the cell open to release DNA along
with other macromolecules such as RNA, proteins,
polysaccharides and also lipids. This can be achieved by treating
the bacterial cells/plant or animal tissue with enzymes such as
lysozyme (bacteria), cellulase (plant cells), chitinase (fungus).
You know that genes are located on long molecules of DNA
interwined with proteins such as histones. The RNA can be removed by
treatment with ribonuclease whereas proteins can be removed by
treatment with protease. Other molecules can be removed by appropriate
treatments and purified DNA ultimately precipitates out after the addition
of chilled ethanol. This can be seen as collection of fine threads in the
suspension (Figure 11.5).

11.3.2 Cutting of DNA at Specific Locations
Restriction enzyme digestions are performed by incubating purified DNA
molecules with the restriction enzyme, at the optimal conditions for that
specific enzyme. Agarose gel electrophoresis is employed to check the
progression of a restriction enzyme digestion. DNA is a negatively charged
molecule, hence it moves towards the positive electrode (anode)
(Figure 11.3). The process is repeated with the vector DNA also.
The joining of DNA involves several processes. After having cut the
source DNA as well as the vector DNA with a specific restriction enzyme,
the cut out ‘gene of interest’ from the source DNA and the cut vector with
space are mixed and ligase is added. This results in the preparation of
recombinant DNA.
11.3.3 Amplification of Gene of Interest using PCR
PCR stands for Polymerase Chain Reaction. In this reaction, multiple
copies of the gene (or DNA) of interest is synthesised in vitro using two
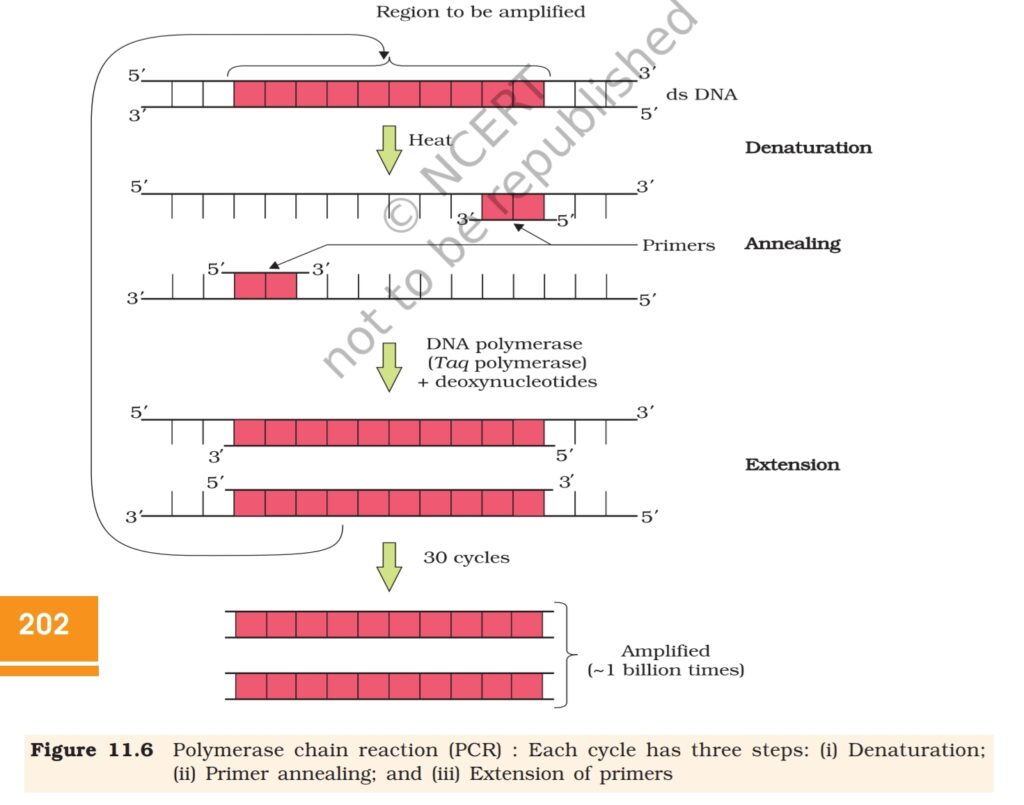
sets of primers (small chemically synthesised oligonucleotides that are
complementary to the regions of DNA) and the enzyme DNA polymerase.
The enzyme extends the primers using the nucleotides provided in the
reaction and the genomic DNA as template. If the process of replication
of DNA is repeated many times, the segment of DNA can be amplified
to approximately billion times, i.e., 1 billion copies are made. Such
repeated amplification is achieved by the use of a thermostable DNA
polymerase (isolated from a bacterium, Thermus aquaticus), which
remain active during the high temperature induced denaturation of
double stranded DNA. The amplified fragment if desired can now be
used to ligate with a vector for further cloning (Figure11.6).
11.3.4 Insertion of Recombinant DNA into the Host
Cell/Organism
There are several methods of introducing the ligated DNA into recipient
cells. Recipient cells after making them ‘competent’ to receive, take up
DNA present in its surrounding. So, if a recombinant DNA bearing gene
for resistance to an antibiotic (e.g., ampicillin) is transferred into E. coli
cells, the host cells become transformed into ampicillin-resistant cells. If
we spread the transformed cells on agar plates containing ampicillin, only
transformants will grow, untransformed recipient cells will die. Since, due
to ampicillin resistance gene, one is able to select a transformed cell in the
presence of ampicillin. The ampicillin resistance gene in this case is called
a selectable marker.
11.3.5 Obtaining the Foreign Gene Product
When you insert a piece of alien DNA into a cloning vector and transfer it
into a bacterial, plant or animal cell, the alien DNA gets multiplied. In
almost all recombinant technologies, the ultimate aim is to produce a
desirable protein. Hence, there is a need for the recombinant DNA to be
expressed. The foreign gene gets expressed under appropriate conditions.
The expression of foreign genes in host cells involve understanding many
technical details.
After having cloned the gene of interest and having optimised the
conditions to induce the expression of the target protein, one has to
consider producing it on a large scale. Can you think of any reason
why there is a need for large-scale production? If any protein encoding
gene is expressed in a heterologous host, it is called a recombinant
protein. The cells harbouring cloned genes of interest may be grown
on a small scale in the laboratory. The cultures may be used for
extracting the desired protein and then purifying it by using different
separation techniques.
The cells can also be multiplied in a continuous culture system wherein
the used medium is drained out from one side while fresh medium is
added from the other to maintain the cells in their physiologically most
active log/exponential phase. This type of culturing method produces a
larger biomass leading to higher yields of desired protein.
Small volume cultures cannot yield appreciable quantities of products.
To produce in large quantities, the development of bioreactors, where
large volumes (100-1000 litres) of culture can be processed, was required.
Thus, bioreactors can be thought of as vessels in which raw materials are
biologically converted into specific products, individual enzymes, etc.,
using microbial plant, animal or human cells. A bioreactor provides the
optimal conditions for achieving the desired product by providing
optimum growth conditions (temperature, pH, substrate, salts, vitamins,
oxygen).
The most commonly used bioreactors are of stirring type, which are
shown in Figure 11.7.
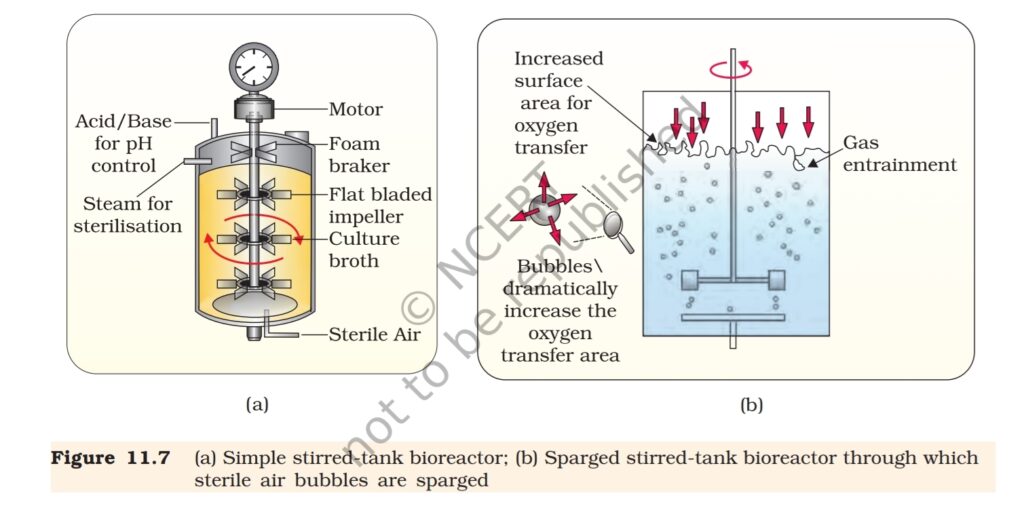
A stirred-tank reactor is usually cylindrical or with a curved base to
facilitate the mixing of the reactor contents. The stirrer facilitates even
mixing and oxygen availability throughout the bioreactor. Alternatively
air can be bubbled through the reactor. If you look at the figure closely
you will see that the bioreactor has an agitator system, an oxygen delivery
system and a foam control system, a temperature control system, pH
control system and sampling ports so that small volumes of the culture
can be withdrawn periodically.
11.3.6 Downstream Processing
After completion of the biosynthetic stage, the product has to be subjected
through a series of processes before it is ready for marketing as a finished
product. The processes include separation and purification, which are
collectively referred to as downstream processing. The product has to be
formulated with suitable preservatives. Such formulation has to undergo
thorough clinical trials as in case of drugs. Strict quality control testing
for each product is also required. The downstream processing and quality
control testing vary from product to product.
SUMMARY
Biotechnology deals with large scale production and marketing of
products and processes using live organisms, cells or enzymes.
Modern biotechnology using genetically modified organisms was
made possible only when man learnt to alter the chemistry of DNA
and construct recombinant DNA. This key process is called
recombinant DNA technology or genetic engineering. This process
involves the use of restriction endonucleases, DNA ligase,
appropriate plasmid or viral vectors to isolate and ferry the foreign
DNA into host organisms, expression of the foreign gene, purification
of the gene product, i.e., the functional protein and finally making a
suitable formulation for marketing. Large scale production involves
use of bioreactors.
EXERCISES
- Can you list 10 recombinant proteins which are used in medical
practice? Find out where they are used as therapeutics (use the internet). - Make a chart (with diagrammatic representation) showing a restriction
enzyme, the substrate DNA on which it acts, the site at which it cuts
DNA and the product it produces. - From what you have learnt, can you tell whether enzymes are bigger or
DNA is bigger in molecular size? How did you know? - What would be the molar concentration of human DNA in a human
cell? Consult your teacher. - Do eukaryotic cells have restriction endonucleases? Justify your answer.
- Besides better aeration and mixing properties, what other advantages
do stirred tank bioreactors have over shake flasks? - Collect 5 examples of palindromic DNA sequences by consulting your teacher.
Better try to create a palindromic sequence by following base-pair rules. - Can you recall meiosis and indicate at what stage a recombinant DNA
is made? - Can you think and answer how a reporter enzyme can be used to monitor
transformation of host cells by foreign DNA in addition to a selectable
marker?
- Describe briefly the following:
(a) Origin of replication
(b) Bioreactors
(c) Downstream processing - Explain briefly
(a) PCR
(b) Restriction enzymes and DNA
(c) Chitinase - Discuss with your teacher and find out how to distinguish between
(a) Plasmid DNA and Chromosomal DNA
(b) RNA and DNA
(c) Exonuclease and Endonuclease
One response to “CHAPTER 11 BIOTECHNOLOGY : PRINCIPLESAND PROCESSES”
This was exactly what I was looking for.