
6.1 The DNA
6.2 The Search for Genetic
Material
6.3 RNA World
6.4 Replication
6.5 Transcription
6.6 Genetic Code
6.7 Translation
6.8 Regulation of Gene
Expression
6.9 Human Genome Project
6.10 DNA Fingerprinting
In the previous chapter, you have learnt the inheritance
patterns and the genetic basis of such patterns. At the
time of Mendel, the nature of those ‘factors’ regulating
the pattern of inheritance was not clear. Over the next
hundred years, the nature of the putative genetic material
was investigated culminating in the realisation that
DNA – deoxyribonucleic acid – is the genetic material, at
least for the majority of organisms. In class XI you have
learnt that nucleic acids are polymers of nucleotides.
Deoxyribonucleic acid (DNA) and ribonucleic acid
(RNA) are the two types of nucleic acids found in living
systems. DNA acts as the genetic material in most of the
organisms. RNA though it also acts as a genetic material
in some viruses, mostly functions as a messenger. RNA
has additional roles as well. It functions as adapter,
structural, and in some cases as a catalytic molecule. In
Class XI you have already learnt the structures of
nucleotides and the way these monomer units are linked
to form nucleic acid polymers. In this chapter we are going
to discuss the structure of DNA, its replication, the process
of making RNA from DNA (transcription), the genetic code
that determines the sequences of amino acids in proteins,
the process of protein synthesis (translation) and
elementary basis of their regulation. The determination
of complete nucleotide sequence of human genome during last decade
has set in a new era of genomics. In the last section, the essentials of
human genome sequencing and its consequences will also be discussed.
Let us begin our discussion by first understanding the structure of
the most interesting molecule in the living system, that is, the DNA. In
subsequent sections, we will understand that why it is the most abundant
genetic material, and what its relationship is with RNA.
6.1 THE DNA
DNA is a long polymer of deoxyribonucleotides. The length of DNA is
usually defined as number of nucleotides (or a pair of nucleotide referred
to as base pairs) present in it. This also is the characteristic of an organism.
For example, a bacteriophage known as φ ×174 has 5386 nucleotides,
Bacteriophage lambda has 48502 base pairs (bp), Escherichia coli has
4.6 × 106
bp, and haploid content of human DNA is 3.3 × 109
bp. Let us
discuss the structure of such a long polymer.
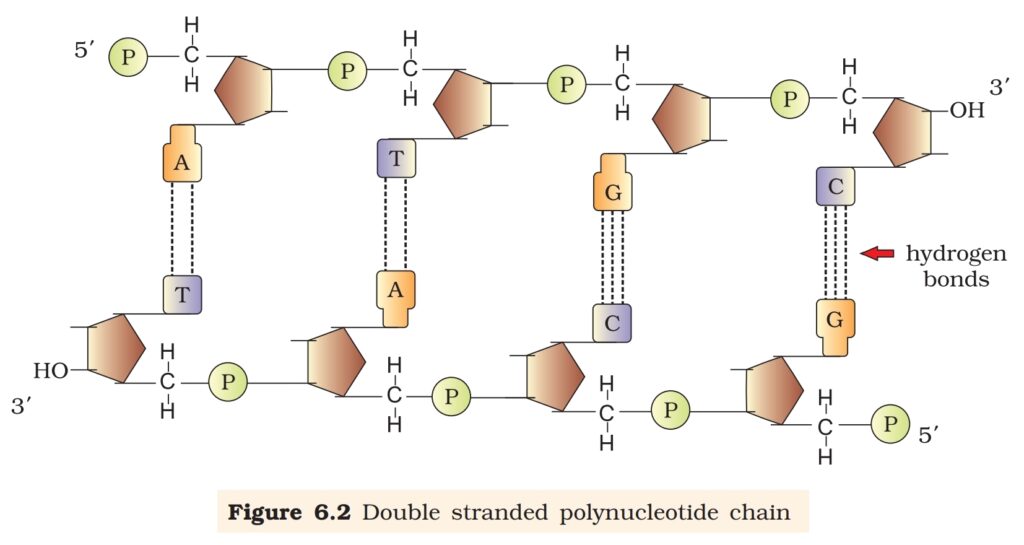
6.1.1 Structure of Polynucleotide Chain
Let us recapitulate the chemical structure of a polynucleotide chain (DNA
or RNA). A nucleotide has three components – a nitrogenous base, a
pentose sugar (ribose in case of RNA, and deoxyribose for DNA), and a
phosphate group. There are two types of nitrogenous bases – Purines
(Adenine and Guanine), and Pyrimidines (Cytosine, Uracil and Thymine).
Cytosine is common for both DNA and RNA and Thymine is present in
DNA. Uracil is present in RNA at the place of Thymine. A nitrogenous
base is linked to the OH of 1′ C pentose sugar through a N-glycosidic
linkage to form a nucleoside, such as adenosine or deoxyadenosine,
guanosine or deoxyguanosine, cytidine or deoxycytidine and uridine or
deoxythymidine. When a phosphate group is linked to OH of 5′ C of a
nucleoside through phosphoester linkage, a corresponding nucleotide
(or deoxynucleotide depending upon the type of sugar present) is formed.
Two nucleotides are linked through 3′-5′ phosphodiester linkage to form
a dinucleotide. More nucleotides can be joined in such a manner to form
a polynucleotide chain. A polymer thus formed has at one end a free

phosphate moiety at 5′ -end of sugar, which is referred to as 5’-end of
polynucleotide chain. Similarly, at the other end of the polymer the sugar
has a free OH of 3’C group which is referred to as 3′ -end of the
polynucleotide chain. The backbone of a polynucleotide chain is formed
due to sugar and phosphates. The nitrogenous bases linked to sugar
moiety project from the backbone (Figure 6.1).
In RNA, every nucleotide residue has an additional –OH group present
at 2′ -position in the ribose. Also, in RNA the uracil is found at the place of
thymine (5-methyl uracil, another chemical name for thymine).
DNA as an acidic substance present in nucleus was first identified by
Friedrich Meischer in 1869. He named it as ‘Nuclein’. However, due to
technical limitation in isolating such a long polymer intact, the elucidation
of structure of DNA remained elusive for a very long period of time. It was
only in 1953 that James Watson and Francis Crick, based on the X-ray
diffraction data produced by Maurice Wilkins and Rosalind Franklin,
proposed a very simple but famous Double Helix model for the structure
of DNA. One of the hallmarks of their proposition was base pairing between
the two strands of polynucleotide chains. However, this proposition was
also based on the observation of Erwin Chargaff that for a double stranded
DNA, the ratios between Adenine and Thymine and Guanine and Cytosine
are constant and equals one.
The base pairing confers a very unique property to the polynucleotide
chains. They are said to be complementary to each other, and therefore if
the sequence of bases in one strand is known then the sequence in other
strand can be predicted. Also, if each strand from a DNA (let us call it as a
parental DNA) acts as a template for synthesis of a new strand, the two
double stranded DNA (let us call them as daughter DNA) thus, produced
would be identical to the parental DNA molecule. Because of this, the genetic
implications of the structure of DNA became very clear.
The salient features of the Double-helix structure of DNA are as follows:
(i) It is made of two polynucleotide chains, where the backbone is
constituted by sugar-phosphate, and the bases project inside.
(ii) The two chains have anti-parallel polarity. It means, if one
chain has the polarity 5’à3′, the other has 3’à5′.
(iii) The bases in two strands are paired through hydrogen bond
(H-bonds) forming base pairs (bp). Adenine forms two hydrogen
bonds with Thymine from opposite strand and vice-versa.
Similarly, Guanine is bonded with Cytosine with three H-bonds.
As a result, always a purine comes opposite to a pyrimidine. This
generates approximately uniform distance between the two
strands of the helix (Figure 6.2).
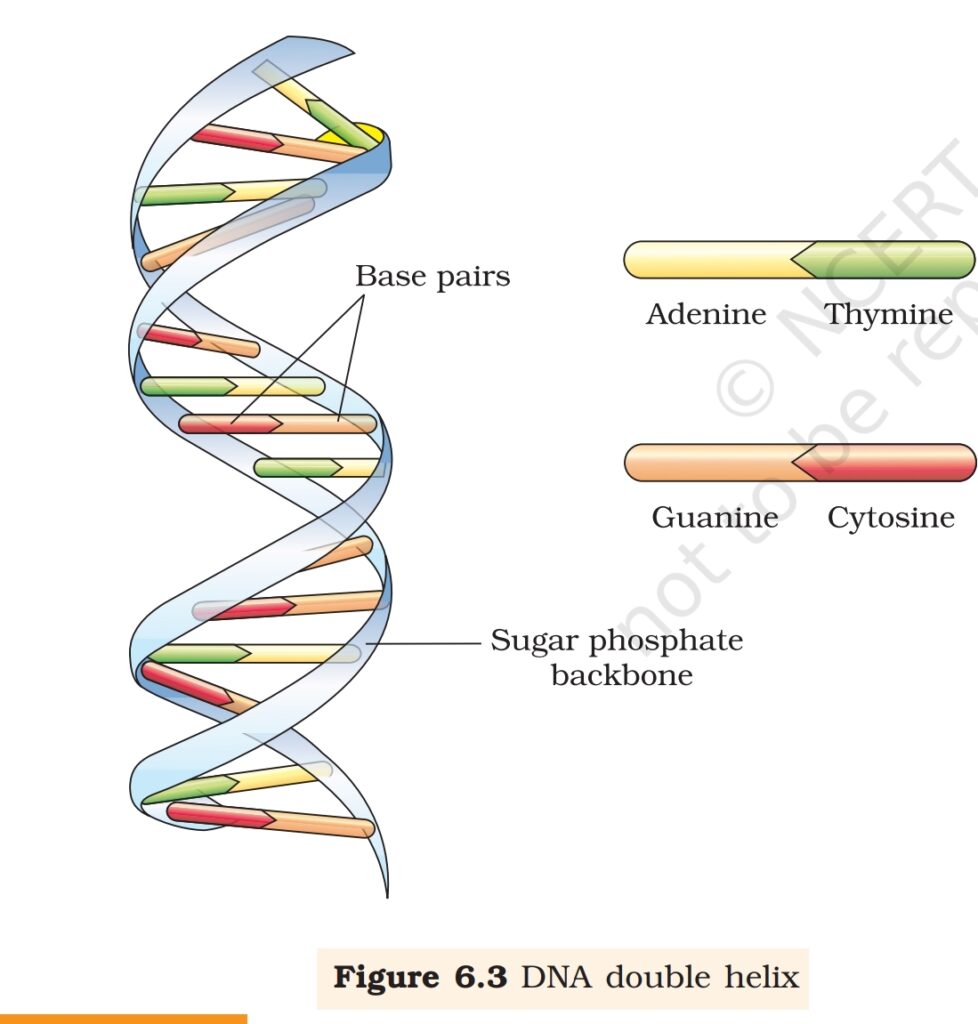
(iv) The two chains are coiled in a right-handed fashion. The pitch
of the helix is 3.4 nm (a nanometre is one billionth of a
metre, that is 10-9 m) and there are roughly 10 bp in each
turn. Consequently, the distance
between a bp in a helix is
approximately 0.34 nm.
(v) The plane of one base pair stacks
over the other in double helix. This,
in addition to H-bonds, confers
stability of the helical structure
(Figure 6.3).
Compare the structure of purines and
pyrimidines. Can you find out why the
distance between two polynucleotide
chains in DNA remains almost constant?
The proposition of a double helix
structure for DNA and its simplicity in
explaining the genetic implication became
revolutionary. Very soon, Francis Crick
proposed the Central dogma in molecular
biology, which states that the genetic
information flows from DNAàRNAàProtein.
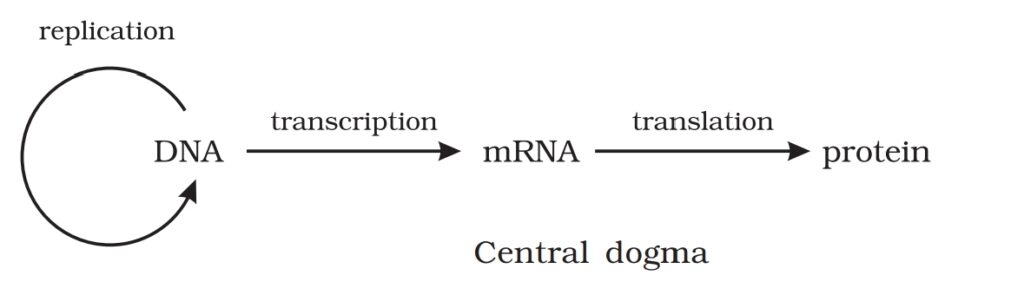
In some viruses the flow of information is in reverse direction, that is,
from RNA to DNA. Can you suggest a simple name to the process?
6.1.2 Packaging of DNA Helix
Taken the distance between two consecutive base pairs
as 0.34 nm (0.34×10–9 m), if the length of DNA double
helix in a typical mammalian cell is calculated (simply
by multiplying the total number of bp with distance
between two consecutive bp, that is, 6.6 × 109 bp ×
0.34 × 10-9m/bp), it comes out to be approximately
2.2 metres. A length that is far greater than the
dimension of a typical nucleus (approximately 10–6 m).
How is such a long polymer packaged in a cell?
If the length of E. coli DNA is 1.36 mm, can you
calculate the number of base pairs in E.coli?
In prokaryotes, such as, E. coli, though they do
not have a defined nucleus, the DNA is not scattered
throughout the cell. DNA (being negatively charged)
is held with some proteins (that have positive
charges) in a region termed as ‘nucleoid’. The DNA
in nucleoid is organised in large loops held by
proteins.
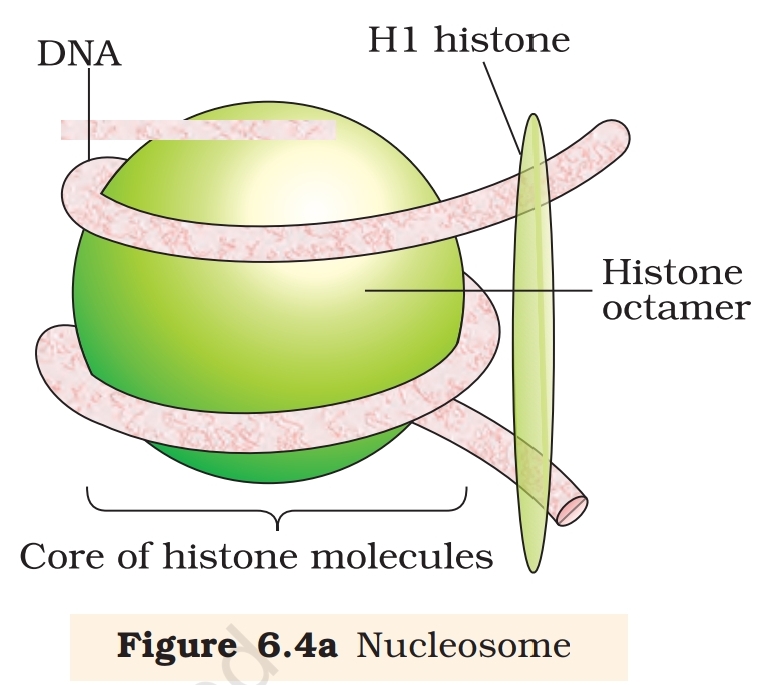
In eukaryotes, this organisation is much more
complex. There is a set of positively charged, basic
proteins called histones. A protein acquires charge
depending upon the abundance of amino acids
residues with charged side chains. Histones are rich
in the basic amino acid residues lysine and arginine.
Both the amino acid residues carry positive charges
in their side chains. Histones are organised to form
a unit of eight molecules called histone octamer.
The negatively charged DNA is wrapped around the positively charged
histone octamer to form a structure called nucleosome (Figure 6.4 a). A
typical nucleosome contains 200 bp of DNA helix. Nucleosomes constitute
the repeating unit of a structure in nucleus called chromatin, thread-
like stained (coloured) bodies seen in nucleus. The nucleosomes in
chromatin are seen as ‘beads-on-string’ structure when viewed under
electron microscope (EM) (Figure 6.4 b).
Theoretically, how many such beads (nucleosomes) do you imagine
are present in a mammalian cell?
The beads-on-string structure in chromatin is packaged to form
chromatin fibers that are further coiled and condensed at metaphase stage
of cell division to form chromosomes. The packaging of chromatin at higher
level requires additional set of proteins that collectively are referred to as

Non-histone Chromosomal (NHC) proteins. In a typical nucleus, some
region of chromatin are loosely packed (and stains light) and are referred to
as euchromatin. The chromatin that is more densely packed and stains
dark are called as Heterochromatin. Euchromatin is said to be
transcriptionally active chromatin, whereas heterochromatin is inactive.
6.2 THE SEARCH FOR GENETIC MATERIAL
Even though the discovery of nuclein by Meischer and the proposition
for principles of inheritance by Mendel were almost at the same time, but
that the DNA acts as a genetic material took long to be discovered and
proven. By 1926, the quest to determine the mechanism for genetic
inheritance had reached the molecular level. Previous discoveries by
Gregor Mendel, Walter Sutton, Thomas Hunt Morgan and numerous other
scientists had narrowed the search to the chromosomes located in the
nucleus of most cells. But the question of what molecule was actually the
genetic material, had not been answered.
Transforming Principle
In 1928, Frederick Griffith, in a series of experiments with Streptococcus
pneumoniae (bacterium responsible for pneumonia), witnessed a
miraculous transformation in the bacteria. During the course of his
experiment, a living organism (bacteria) had changed in physical form.
When Streptococcus pneumoniae (pneumococcus) bacteria are grown
on a culture plate, some produce smooth shiny colonies (S) while others
produce rough colonies (R). This is because the S strain bacteria have a
mucous (polysaccharide) coat, while R strain does not. Mice infected with
the S strain (virulent) die from pneumonia infection but mice infected
with the R strain do not develop pneumonia.

Griffith was able to kill bacteria by heating them. He observed that
heat-killed S strain bacteria injected into mice did not kill them. When he

injected a mixture of heat-killed S and live R bacteria, the mice died.
Moreover, he recovered living S bacteria from the dead mice.
He concluded that the R strain bacteria had somehow been
transformed by the heat-killed S strain bacteria. Some ‘transforming
principle’, transferred from the heat-killed S strain, had enabled the
R strain to synthesise a smooth polysaccharide coat and become virulent.
This must be due to the transfer of the genetic material. However, the
biochemical nature of genetic material was not defined from his
experiments.
Biochemical Characterisation of Transforming Principle
Prior to the work of Oswald Avery, Colin MacLeod and Maclyn McCarty
(1933-44), the genetic material was thought to be a protein. They worked
to determine the biochemical nature of ‘transforming principle’ in Griffith’s
experiment.
They purified biochemicals (proteins, DNA, RNA, etc.) from the
heat-killed S cells to see which ones could transform live R cells into
S cells. They discovered that DNA alone from S bacteria caused R bacteria
to become transformed.
They also discovered that protein-digesting enzymes (proteases) and
RNA-digesting enzymes (RNases) did not affect transformation, so the
transforming substance was not a protein or RNA. Digestion with DNase
did inhibit transformation, suggesting that the DNA caused the
transformation. They concluded that DNA is the hereditary material, but
not all biologists were convinced.
Can you think of any difference between DNAs and DNase?
6.2.1 The Genetic Material is DNA
The unequivocal proof that DNA is the genetic material came from the
experiments of Alfred Hershey and Martha Chase (1952). They worked
with viruses that infect bacteria called bacteriophages.
The bacteriophage attaches to the bacteria and its genetic material
then enters the bacterial cell. The bacterial cell treats the viral genetic
material as if it was its own and subsequently manufactures more virus
particles. Hershey and Chase worked to discover whether it was protein
or DNA from the viruses that entered the bacteria.
They grew some viruses on a medium that contained radioactive
phosphorus and some others on medium that contained radioactive sulfur.
Viruses grown in the presence of radioactive phosphorus contained
radioactive DNA but not radioactive protein because DNA contains
phosphorus but protein does not. Similarly, viruses grown on radioactive
sulfur contained radioactive protein but not radioactive DNA because
DNA does not contain sulfur.
Radioactive phages were allowed to attach to E. coli bacteria. Then, as
the infection proceeded, the viral coats were removed from the bacteria by
agitating them in a blender. The virus particles were separated from the
bacteria by spinning them in a centrifuge.
Bacteria which was infected with viruses that had radioactive DNA
were radioactive, indicating that DNA was the material that passed from
the virus to the bacteria. Bacteria that were infected with viruses that had
radioactive proteins were not radioactive. This indicates that proteins did
not enter the bacteria from the viruses. DNA is therefore the genetic
material that is passed from virus to bacteria (Figure 6.5).

6.2.2 Properties of Genetic Material (DNA versus RNA)
From the foregoing discussion, it is clear that the debate between proteins
versus DNA as the genetic material was unequivocally resolved from
Hershey-Chase experiment. It became an established fact that it is DNA
that acts as genetic material. However, it subsequently became clear that
in some viruses, RNA is the genetic material (for example, Tobacco Mosaic
viruses, QB bacteriophage, etc.). Answer to some of the questions such as,
why DNA is the predominant genetic material, whereas RNA performs
dynamic functions of messenger and adapter has to be found from the
differences between chemical structures of the two nucleic acid molecules.
Can you recall the two chemical differences between DNA and RNA?
A molecule that can act as a genetic material must fulfill the following
criteria:
(i) It should be able to generate its replica (Replication).
(ii) It should be stable chemically and structurally.
(iii) It should provide the scope for slow changes (mutation) that
are required for evolution.
(iv) It should be able to express itself in the form of ‘Mendelian
Characters’.
If one examines each requirement one by one, because of rule of base
pairing and complementarity, both the nucleic acids (DNA and RNA) have
the ability to direct their duplications. The other molecules in the living
system, such as proteins fail to fulfill first criteria itself.
The genetic material should be stable enough not to change with
different stages of life cycle, age or with change in physiology of the
organism. Stability as one of the properties of genetic material was very
evident in Griffith’s ‘transforming principle’ itself that heat, which killed
the bacteria, at least did not destroy some of the properties of genetic
material. This now can easily be explained in light of the DNA that the
two strands being complementary if separated by heating come together,
when appropriate conditions are provided. Further, 2′-OH group present
at every nucleotide in RNA is a reactive group and makes RNA labile and
easily degradable. RNA is also now known to be catalytic, hence reactive.
Therefore, DNA chemically is less reactive and structurally more stable
when compared to RNA. Therefore, among the two nucleic acids, the DNA
is a better genetic material.
In fact, the presence of thymine at the place of uracil also confers
additional stability to DNA. (Detailed discussion about this requires
understanding of the process of repair in DNA, and you will study these
processes in higher classes.)
Both DNA and RNA are able to mutate. In fact, RNA being unstable,
mutate at a faster rate. Consequently, viruses having RNA genome and
having shorter life span mutate and evolve faster.
RNA can directly code for the synthesis of proteins, hence can easily
express the characters. DNA, however, is dependent on RNA for synthesis
of proteins. The protein synthesising machinery has evolved around RNA.
The above discussion indicate that both RNA and DNA can function as
genetic material, but DNA being more stable is preferred for storage of
genetic information. For the transmission of genetic information, RNA
is better.
6.3 RNA WORLD
From foregoing discussion, an immediate question becomes evident –
which is the first genetic material? It shall be discussed in detail in the
chapter on chemical evolution, but briefly, we shall highlight some of the
facts and points.
RNA was the first genetic material. There is now enough evidence to
suggest that essential life processes (such as metabolism, translation,
splicing, etc.), evolved around RNA. RNA used to act as
a genetic material as well as a catalyst (there are some
important biochemical reactions in living systems that
are catalysed by RNA catalysts and not by protein
enzymes). But, RNA being a catalyst was reactive and
hence unstable. Therefore, DNA has evolved from RNA
with chemical modifications that make it more stable.
DNA being double stranded and having complementary
strand further resists changes by evolving a process of
repair.
6.4 REPLICATION
While proposing the double helical structure for DNA,
Watson and Crick had immediately proposed a scheme
for replication of DNA. To quote their original statement
that is as follows:
‘‘It has not escaped our notice that the specific
pairing we have postulated immediately suggests a
possible copying mechanism for the genetic material’’
(Watson and Crick, 1953).
The scheme suggested that the two strands would
separate and act as a template for the synthesis of new
complementary strands. After the completion of
replication, each DNA molecule would have one
parental and one newly synthesised strand. This
scheme was termed as semiconservative DNA
replication (Figure 6.6).

6.4.1 The Experimental Proof
It is now proven that DNA replicates semiconservatively. It was shown first in
Escherichia coli and subsequently in higher organisms, such as plants
and human cells. Matthew Meselson and Franklin Stahl performed the
following experiment in 1958:
(i) They grew E. coli in a medium containing 15NH4Cl (15N is the heavy
isotope of nitrogen) as the only nitrogen source for many
generations. The result was that 15N was incorporated into newly
synthesised DNA (as well as other nitrogen containing compounds).
This heavy DNA molecule could be distinguished from the normal
DNA by centrifugation in a cesium chloride (CsCl) density gradient
(Please note that 15N is not a radioactive isotope, and it can be
separated from 14N only based on densities).
(ii) Then they transferred the cells into a medium with normal
14NH4Cl and took samples at various definite time intervals as
the cells multiplied, and extracted the DNA that remained as
double-stranded helices. The various samples were separated
independently on CsCl gradients to measure the densities of
DNA (Figure 6.7).
Can you recall what centrifugal force is, and think why a
molecule with higher mass/density would sediment faster?
The results are shown in Figure 6.7.

(iii) Thus, the DNA that was extracted from the culture one
generation after the transfer from 15N to 14N medium [that is
after 20 minutes; E. coli divides in 20 minutes] had a hybrid or
intermediate density. DNA extracted from the culture after
another generation [that is after 40 minutes, II generation] was
composed of equal amounts of this hybrid DNA and of ‘light’
DNA.
If E. coli was allowed to grow for 80 minutes then what would be the
proportions of light and hybrid densities DNA molecule?
Very similar experiments involving use of radioactive thymidine to
detect distribution of newly synthesised DNA in the chromosomes was
performed on Vicia faba (faba beans) by Taylor and colleagues in 1958.
The experiments proved that the DNA in chromosomes also replicate
semiconservatively.
6.4.2 The Machinery and the Enzymes
In living cells, such as E. coli, the process of replication requires a set of
catalysts (enzymes). The main enzyme is referred to as DNA-dependent
DNA polymerase, since it uses a DNA template to catalyse the
polymerisation of deoxynucleotides. These enzymes are highly efficient
enzymes as they have to catalyse polymerisation of a large number of
nucleotides in a very short time. E. coli that has only 4.6 ×106
bp (compare
it with human whose diploid content is 6.6 × 109
bp), completes the
process of replication within 18 minutes; that means the average rate of
polymerisation has to be approximately 2000 bp per second. Not only do
these polymerases have to be fast, but they also have to catalyse the reaction
with high degree of accuracy. Any mistake during replication would result
into mutations. Furthermore, energetically replication is a very expensive
process. Deoxyribonucleoside triphosphates serve dual purposes. In
addition to acting as substrates, they provide energy for polymerisation
reaction (the two terminal phosphates in a deoxynucleoside triphosphates
are high-energy phosphates, same as in case of ATP).
In addition to DNA-dependent DNA polymerases, many additional
enzymes are required to complete the process of replication with high
degree of accuracy. For long DNA molecules, since the two strands of
DNA cannot be separated in its entire length (due to very high energy
requirement), the replication occur within a small opening of the DNA
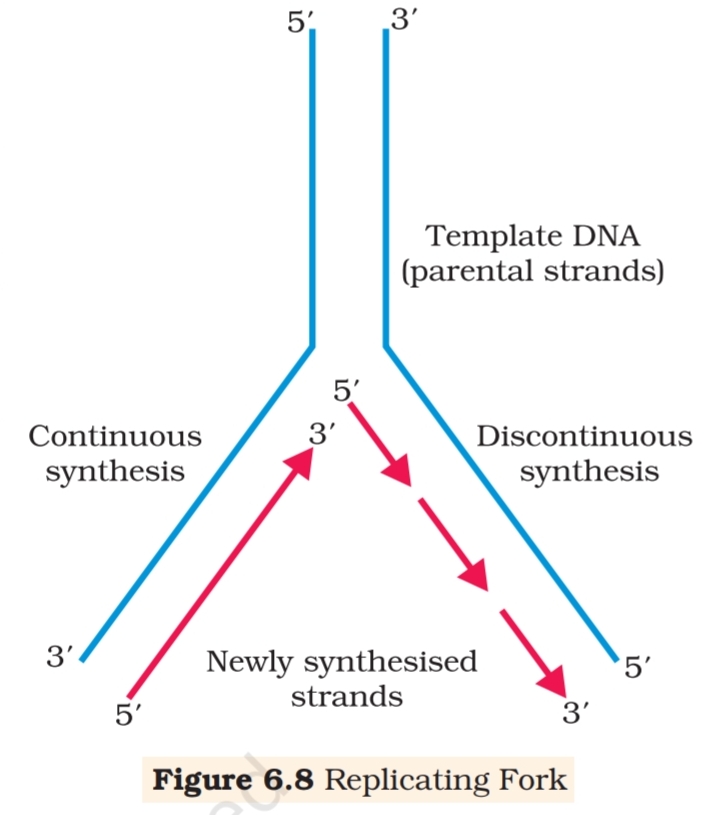
helix, referred to as replication fork. The DNA-dependent DNA
polymerases catalyse polymerisation only in one direction, that is 5’à3′.
This creates some additional complications at the replicating fork.
Consequently, on one strand (the template with polarity 3’à5′), the
replication is continuous, while on the other (the template with
polarity 5’à3′), it is discontinuous. The discontinuously synthesised
fragments are later joined by the enzyme DNA ligase (Figure 6.8).
The DNA polymerases on their own cannot initiate the process of
replication. Also the replication does not initiate randomly at any place
in DNA. There is a definite region in E. coli DNA where the replication
originates. Such regions are termed as origin of replication. It is
because of the requirement of the origin of
replication that a piece of DNA if needed to be
propagated during recombinant DNA procedures,
requires a vector. The vectors provide the origin of
replication.
Further, not every detail of replication is
understood well. In eukaryotes, the replication of
DNA takes place at S-phase of the cell-cycle. The
replication of DNA and cell division cycle should be
highly coordinated. A failure in cell division after
DNA replication results into polyploidy(a
chromosomal anomaly). You will learn the detailed
nature of origin and the processes occurring at this
site, in higher classes.
6.5 TRANSCRIPTION
The process of copying genetic information from one
strand of the DNA into RNA is termed as
transcription. Here also, the principle of
complementarity governs the process of transcription, except the adenosine
complements now forms base pair with uracil instead of thymine. However,
unlike in the process of replication, which once set in, the total DNA of an
organism gets duplicated, in transcription only a segment of DNA and
only one of the strands is copied into RNA. This necessitates defining the
boundaries that would demarcate the region and the strand of DNA that
would be transcribed.
Why both the strands are not copied during transcription has the
simple answer. First, if both strands act as a template, they would code
for RNA molecule with different sequences (Remember complementarity
does not mean identical), and in turn, if they code for proteins, the sequence
of amino acids in the proteins would be different. Hence, one segment of
the DNA would be coding for two different proteins, and this would
complicate the genetic information transfer machinery. Second, the two
RNA molecules if produced simultaneously would be complementary to
each other, hence would form a double stranded RNA. This would prevent
RNA from being translated into protein and the exercise of transcription
would become a futile one.
6.5.1 Transcription Unit
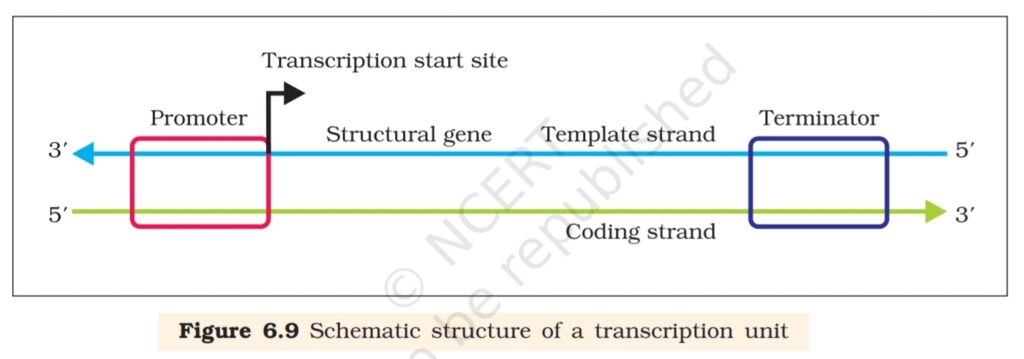
A transcription unit in DNA is defined primarily by the three regions in
the DNA:
(i) A Promoter
(ii) The Structural gene
(iii) A Terminator
There is a convention in defining the two strands of the DNA in the
structural gene of a transcription unit. Since the two strands have opposite
polarity and the DNA-dependent RNA polymerase also catalyse the
polymerisation in only one direction, that is, 5’→3′, the strand that has
the polarity 3’→5′ acts as a template, and is also referred to as template
strand. The other strand which has the polarity (5’→3′) and the sequence
same as RNA (except thymine at the place of uracil), is displaced during
transcription. Strangely, this strand (which does not code for anything)
is referred to as coding strand. All the reference point while defining a
transcription unit is made with coding strand. To explain the point, a
hypothetical sequence from a transcription unit is represented below:
3′ -ATGCATGCATGCATGCATGCATGC-5′ Template Strand
5′ -TACGTACGTACGTACGTACGTACG-3′ Coding Strand
Can you now write the sequence of RNA transcribed from the above DNA?
The promoter and terminator flank the structural gene in a
transcription unit. The promoter is said to be located towards 5′ -end
(upstream) of the structural gene (the reference is made with respect to
the polarity of coding strand). It is a DNA sequence that provides binding
site for RNA polymerase, and it is the presence of a promoter in a
transcription unit that also defines the template and coding strands. By
switching its position with terminator, the definition of coding and template
strands could be reversed. The terminator is located towards 3′ -end
(downstream) of the coding strand and it usually defines the end of the
process of transcription (Figure 6.9). There are additional regulatory
sequences that may be present further upstream or downstream to the
promoter. Some of the properties of these sequences shall be discussed
while dealing with regulation of gene expression.
6.5.2 Transcription Unit and the Gene
A gene is defined as the functional unit of inheritance. Though there is no
ambiguity that the genes are located on the DNA, it is difficult to literally
define a gene in terms of DNA sequence. The DNA sequence coding for
tRNA or rRNA molecule also define a gene. However by defining a cistron
as a segment of DNA coding for a polypeptide, the structural gene in a
transcription unit could be said as monocistronic (mostly in eukaryotes)
or polycistronic (mostly in bacteria or prokaryotes). In eukaryotes, the
monocistronic structural genes have interrupted coding sequences – the
genes in eukaryotes are split. The coding sequences or expressed
sequences are defined as exons. Exons are said to be those sequence
that appear in mature or processed RNA. The exons are interrupted by
introns. Introns or intervening sequences do not appear in mature or
processed RNA. The split-gene arrangement further complicates the
definition of a gene in terms of a DNA segment.
Inheritance of a character is also affected by promoter and regulatory
sequences of a structural gene. Hence, sometime the regulatory sequences
are loosely defined as regulatory genes, even though these sequences do
not code for any RNA or protein.
6.5.3 Types of RNA and the process of Transcription
In bacteria, there are three major types of RNAs: mRNA (messenger RNA),
tRNA (transfer RNA), and rRNA (ribosomal RNA). All three RNAs are
needed to synthesise a protein in a cell. The mRNA provides the template,
tRNA brings aminoacids and reads the genetic code, and rRNAs play
structural and catalytic role during translation. There is single
DNA-dependent RNA polymerase that catalyses transcription of all types
of RNA in bacteria. RNA polymerase binds to promoter and initiates
transcription (Initiation). It uses nucleoside triphosphates as substrate
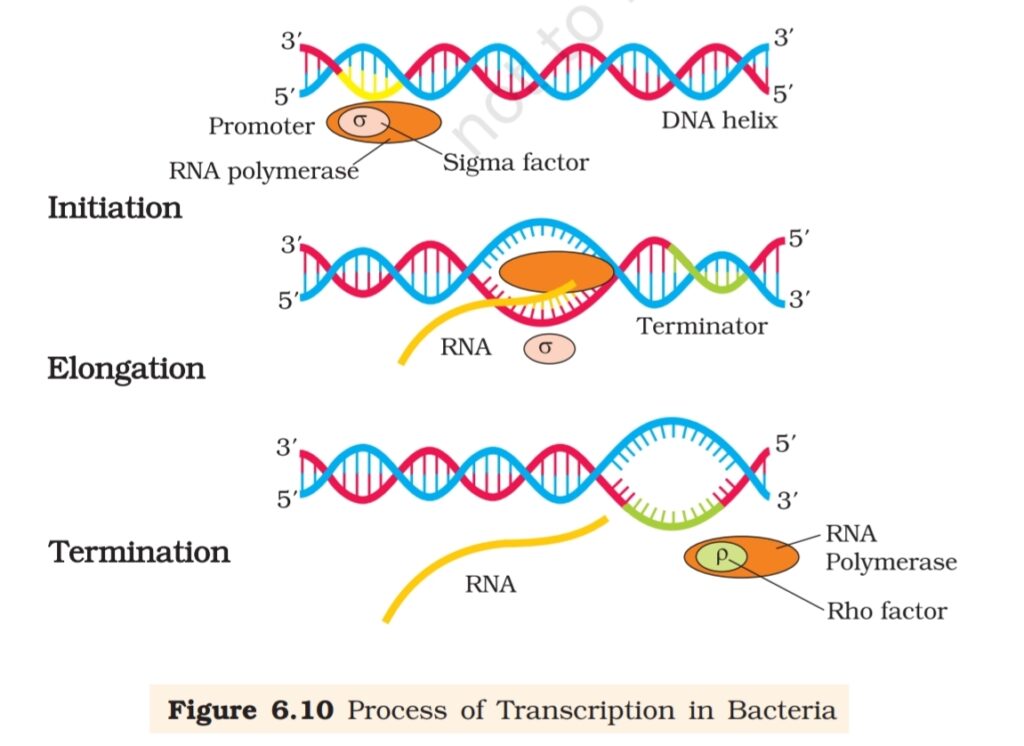
and polymerises in a template depended fashion following the rule of
complementarity. It somehow also facilitates opening of the helix and
continues elongation. Only a short stretch of RNA remains bound to the
enzyme. Once the polymerases reaches the terminator region, the nascent
RNA falls off, so also the RNA polymerase. This results in termination of
transcription.
An intriguing question is that how is the RNA polymerases able
to catalyse all the three steps, which are initiation, elongation and
termination. The RNA polymerase is only capable of catalysing the
process of elongation. It associates transiently with initiation-factor (σ)
and termination-factor (ρ) to initiate and terminate the transcription,
respectively. Association with these factors alter the specificity of the
RNA polymerase to either initiate or terminate (Figure 6.10).
In bacteria, since the mRNA does not require any processing to become
active, and also since transcription and translation take place in the same
compartment (there is no separation of cytosol and nucleus in bacteria),
many times the translation can begin much before the mRNA is fully
transcribed. Consequently, the transcription and translation can be coupled
in bacteria.
In eukaryotes, there are two additional complexities –
(i) There are at least three RNA polymerases in the nucleus (in addition
to the RNA polymerase found in the organelles). There is a clear
cut division of labour. The RNA polymerase I transcribes rRNAs
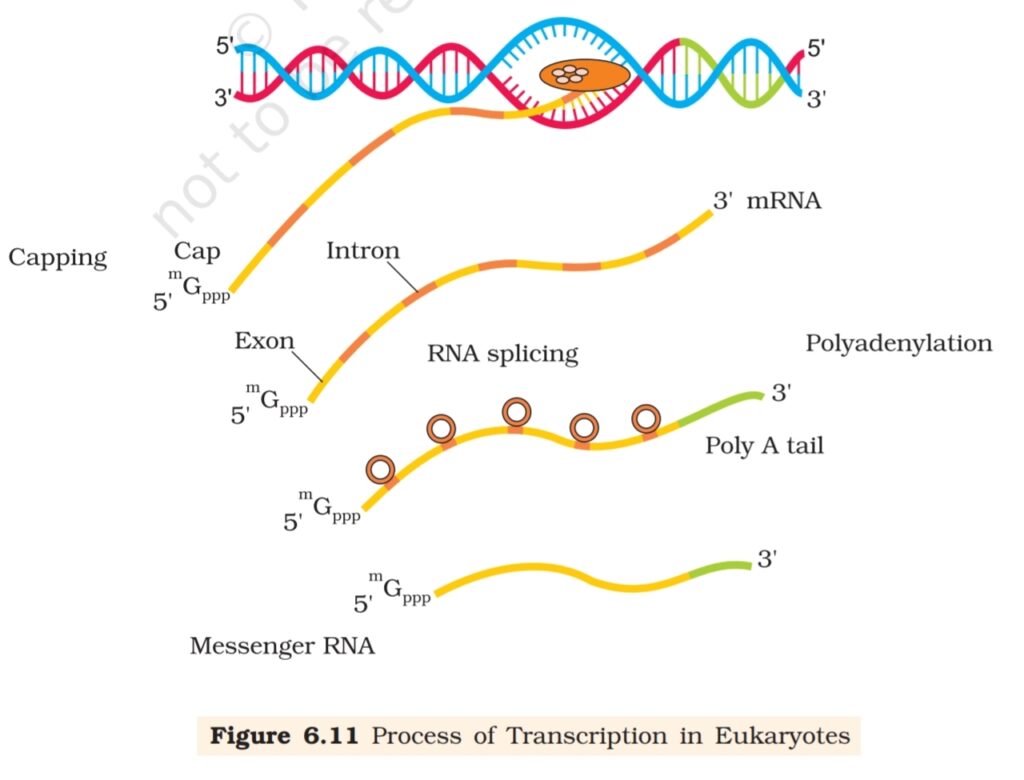
(28S, 18S, and 5.8S), whereas the RNA polymerase III is responsible
for transcription of tRNA, 5srRNA, and snRNAs (small nuclear
RNAs). The RNA polymerase II transcribes precursor of mRNA, the
heterogeneous nuclear RNA (hnRNA).
(ii) The second complexity is that the primary transcripts contain both
the exons and the introns and are non-functional. Hence, it is
subjected to a process called splicing where the introns are removed
and exons are joined in a defined order. hnRNA undergoes
additional processing called as capping and tailing. In capping an
unusual nucleotide (methyl guanosine triphosphate) is added to
the 5′-end of hnRNA. In tailing, adenylate residues (200-300) are
added at 3′-end in a template independent manner. It is the fully
processed hnRNA, now called mRNA, that is transported out of the
nucleus for translation (Figure 6.11).
The significance of such complexities is now beginning to be
understood. The split-gene arrangements represent probably an ancient
feature of the genome. The presence of introns is reminiscent of antiquity,
and the process of splicing represents the dominance of RNA-world. In
recent times, the understanding of RNA and RNA-dependent processes
in the living system have assumed more importance.
6.6 GENETIC CODE
During replication and transcription a nucleic acid was copied to form
another nucleic acid. Hence, these processes are easy to conceptualise
on the basis of complementarity. The process of translation requires
transfer of genetic information from a polymer of nucleotides to synthesise
a polymer of amino acids. Neither does any complementarity exist between
nucleotides and amino acids, nor could any be drawn theoretically. There
existed ample evidences, though, to support the notion that change in
nucleic acids (genetic material) were responsible for change in amino acids
in proteins. This led to the proposition of a genetic code that could direct
the sequence of amino acids during synthesis of proteins.
If determining the biochemical nature of genetic material and the
structure of DNA was very exciting, the proposition and deciphering of
genetic code were most challenging. In a very true sense, it required
involvement of scientists from several disciplines – physicists, organic
chemists, biochemists and geneticists. It was George Gamow, a physicist,
who argued that since there are only 4 bases and if they have to code for
20 amino acids, the code should constitute a combination of bases. He
suggested that in order to code for all the 20 amino acids, the code should
be made up of three nucleotides. This was a very bold proposition, because
a permutation combination of 43
(4 × 4 × 4) would generate 64 codons;
generating many more codons than required.
Providing proof that the codon was a triplet, was a more daunting
task. The chemical method developed by Har Gobind Khorana was
instrumental in synthesising RNA molecules with defined combinations
of bases (homopolymers and copolymers). Marshall Nirenberg’s cell-free
system for protein synthesis finally helped the code to be deciphered.
Severo Ochoa enzyme (polynucleotide phosphorylase) was also helpful
in polymerising RNA with defined sequences in a template independent
manner (enzymatic synthesis of RNA). Finally a checker-board for genetic
code was prepared which is given in Table 6.1.
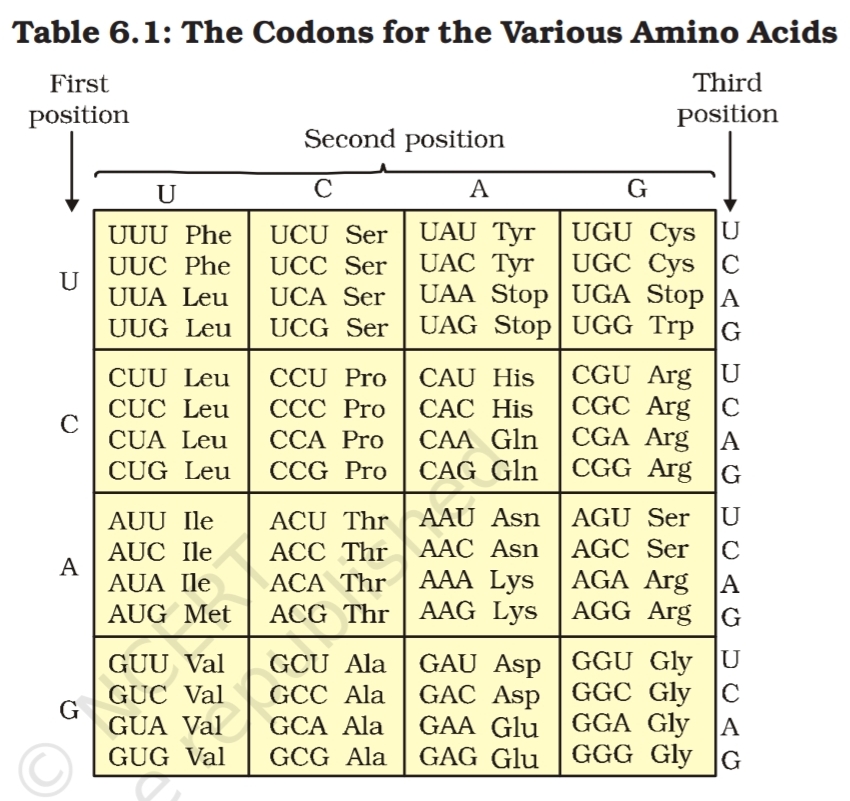
The salient features of genetic code are as follows:
(i) The codon is triplet. 61 codons code for amino acids and 3 codons do
not code for any amino acids, hence they function as stop codons.
(ii) Some amino acids are coded by more than one codon, hence
the code is degenerate.
(iii) The codon is read in mRNA in a contiguous fashion. There are
no punctuations.
(iv) The code is nearly universal: for example, from bacteria to human
UUU would code for Phenylalanine (phe). Some exceptions to this
rule have been found in mitochondrial codons, and in some
protozoans.
(v) AUG has dual functions. It codes for Methionine (met) , and it
also act as initiator codon.
(vi) UAA, UAG, UGA are stop terminator codons.
If following is the sequence of nucleotides in mRNA, predict the
sequence of amino acid coded by it (take help of the checkerboard):
-AUG UUU UUC UUC UUU UUU UUC-
Now try the opposite. Following is the sequence of amino acids coded
by an mRNA. Predict the nucleotide sequence in the RNA:
Met-Phe-Phe-Phe-Phe-Phe-Phe
Do you face any difficulty in predicting the opposite?
Can you now correlate which two properties of genetic code you have
learnt?
6.6.1 Mutations and Genetic Code
The relationships between genes and DNA are best understood by mutation
studies. You have studied about mutation and its effect in Chapter 5. Effects
of large deletions and rearrangements in a segment of DNA are easy to
comprehend. It may result in loss or gain of a gene and so a function. The
effect of point mutations will be explained here. A classical example of
point mutation is a change of single base pair in the gene for beta globin
chain that results in the change of amino acid residue glutamate to valine.
It results into a diseased condition called as sickle cell anemia. Effect of
point mutations that inserts or deletes a base in structural gene can be
better understood by following simple example.
Consider a statement that is made up of the following words each
having three letters like genetic code.
RAM HAS RED CAP
If we insert a letter B in between HAS and RED and rearrange the
statement, it would read as follows:
RAM HAS BRE DCA P
Similarly, if we now insert two letters at the same place, say BI’. Now it
would read,
RAM HAS BIR EDC AP
Now we insert three letters together, say BIG, the statement would read
RAM HAS BIG RED CAP
The same exercise can be repeated, by deleting the letters R, E and D,
one by one and rearranging the statement to make a triplet word.
RAM HAS EDC AP
RAM HAS DCA P
RAM HAS CAP
The conclusion from the above exercise is very obvious. Insertion or
deletion of one or two bases changes the reading frame from the point of
insertion or deletion. However, such mutations are referred to as
frameshift insertion or deletion mutations. Insertion or deletion of
three or its multiple bases insert or delete in one or multiple codon hence
one or multiple amino acids, and reading frame remains unaltered from
that point onwards.
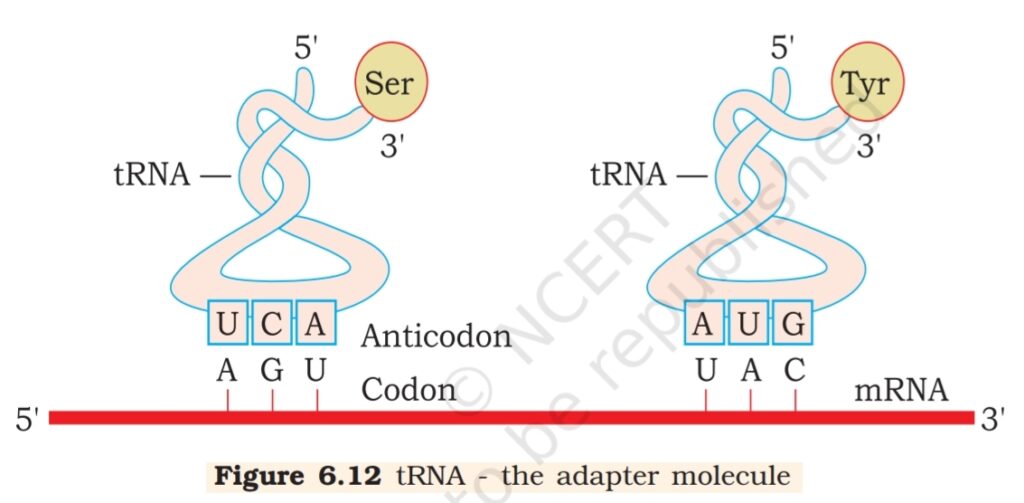
6.6.2 tRNA– the Adapter Molecule
From the very beginning of the proposition of code, it was clear to Francis
Crick that there has to be a mechanism to read the code and also to link it
to the amino acids, because amino acids have no structural specialities to
read the code uniquely. He postulated the presence of an adapter molecule
that would on one hand read the code and on other hand would bind
to specific amino acids. The tRNA, then called sRNA (soluble RNA),
was known before the genetic code was postulated. However, its role
as an adapter molecule was assigned much later.
tRNA has an
anticodon loop
that has bases
complementary to
the code, and it also
has an amino acid
acceptor end to
which it binds to
amino acids.
tRNAs are specific
for each amino acid
(Figure 6.12). For
initiation, there is
another specific tRNA that is referred to as initiator tRNA. There are no
tRNAs for stop codons. In figure 6.12, the secondary structure of tRNA
has been depicted that looks like a clover-leaf. In actual structure, the
tRNA is a compact molecule which looks like inverted L.
6.7 TRANSLATION
Translation refers to the process of polymerisation of amino acids to
form a polypeptide (Figure 6.13). The order and sequence of amino acids
are defined by the sequence of bases in the mRNA. The amino acids are
joined by a bond which is known as a peptide bond. Formation of a
peptide bond requires energy. Therefore, in the first phase itself amino
acids are activated in the presence of ATP and linked to their cognate
tRNA – a process commonly called as charging of tRNA or
aminoacylation of tRNA to be more specific. If two such charged tRNAs
are brought close enough, the formation of peptide bond between them
would be favoured energetically. The
presence of a catalyst would enhance
the rate of peptide bond formation.
The cellular factory responsible for
synthesising proteins is the ribosome.
The ribosome consists of structural
RNAs and about 80 different proteins.
In its inactive state, it exists as two
subunits; a large subunit and a small
subunit. When the small subunit
encounters an mRNA, the process of
translation of the mRNA to protein
begins. There are two sites in the large
subunit, for subsequent amino acids
to bind to and thus, be close enough
to each other for the formation of a
peptide bond. The ribosome also acts as a catalyst (23S rRNA in bacteria
is the enzyme- ribozyme) for the formation of peptide bond.
A translational unit in mRNA is the sequence of RNA that is flanked
by the start codon (AUG) and the stop codon and codes for a polypeptide.
An mRNA also has some additional sequences that are not translated
and are referred as untranslated regions (UTR). The UTRs are present
at both 5′ -end (before start codon) and at 3′ -end (after stop codon). They
are required for efficient translation process.

For initiation, the ribosome binds to the mRNA at the start codon (AUG)
that is recognised only by the initiator tRNA. The ribosome proceeds to the
elongation phase of protein synthesis. During this stage, complexes
composed of an amino acid linked to tRNA, sequentially bind to the
appropriate codon in mRNA by forming complementary base pairs with
the tRNA anticodon. The ribosome moves from codon to codon along the
mRNA. Amino acids are added one by one, translated into Polypeptide
sequences dictated by DNA and represented by mRNA. At the end, a release
factor binds to the stop codon, terminating translation and releasing the
complete polypeptide from the ribosome.
6.8 REGULATION OF GENE EXPRESSION
Regulation of gene expression refers to a very broad term that may occur
at various levels. Considering that gene expression results in the formation
of a polypeptide, it can be regulated at several levels. In eukaryotes, the
regulation could be exerted at
(i) transcriptional level (formation of primary transcript),
(ii) processing level (regulation of splicing),
(iii) transport of mRNA from nucleus to the cytoplasm,
(iv) translational level.
The genes in a cell are expressed to perform a particular function or a
set of functions. For example, if an enzyme called beta-galactosidase is
synthesised by E. coli, it is used to catalyse the hydrolysis of a
disaccharide, lactose into galactose and glucose; the bacteria use them
as a source of energy. Hence, if the bacteria do not have lactose around
them to be utilised for energy source, they would no longer require the
synthesis of the enzyme beta-galactosidase. Therefore, in simple terms,
it is the metabolic, physiological or environmental conditions that regulate
the expression of genes. The development and differentiation of embryo
into adult organisms are also a result of the coordinated regulation of
expression of several sets of genes.
In prokaryotes, control of the rate of transcriptional initiation is the
predominant site for control of gene expression. In a transcription unit,
the activity of RNA polymerase at a given promoter is in turn regulated
by interaction with accessory proteins, which affect its ability to recognise
start sites. These regulatory proteins can act both positively (activators)
and negatively (repressors). The accessibility of promoter regions of
prokaryotic DNA is in many cases regulated by the interaction of proteins
with sequences termed operators. The operator region is adjacent to the
promoter elements in most operons and in most cases the sequences of
the operator bind a repressor protein. Each operon has its specific
operator and specific repressor. For example, lac operator is present
only in the lac operon and it interacts specifically with lac repressor only.
6.8.1 The Lac operon
The elucidation of the lac operon was also a result of a close association
between a geneticist, Francois Jacob and a biochemist, Jacque Monod. They
were the first to elucidate a transcriptionally regulated system. In lac operon
(here lac refers to lactose), a polycistronic structural gene is regulated by a
common promoter and regulatory genes. Such arrangement is very common
in bacteria and is referred to as operon. To name few such examples, lac
operon, trp operon, ara operon, his operon, val operon, etc.
The lac operon consists of one regulatory gene (the i gene – here the
term i does not refer to inducer, rather it is derived from the word inhibitor)
and three structural genes (z, y, and a). The i gene codes for the repressor
of the lac operon. The z gene codes for beta-galactosidase (β-gal), which
is primarily responsible for the hydrolysis of the disaccharide, lactose
into its monomeric units, galactose and glucose. The y gene codes for
permease, which increases permeability of the cell to β-galactosides. The
a gene encodes a transacetylase. Hence, all the three gene products in
lac operon are required for metabolism of lactose. In most other operons
as well, the genes present in the operon are needed together to function
in the same or related metabolic pathway (Figure 6.14).
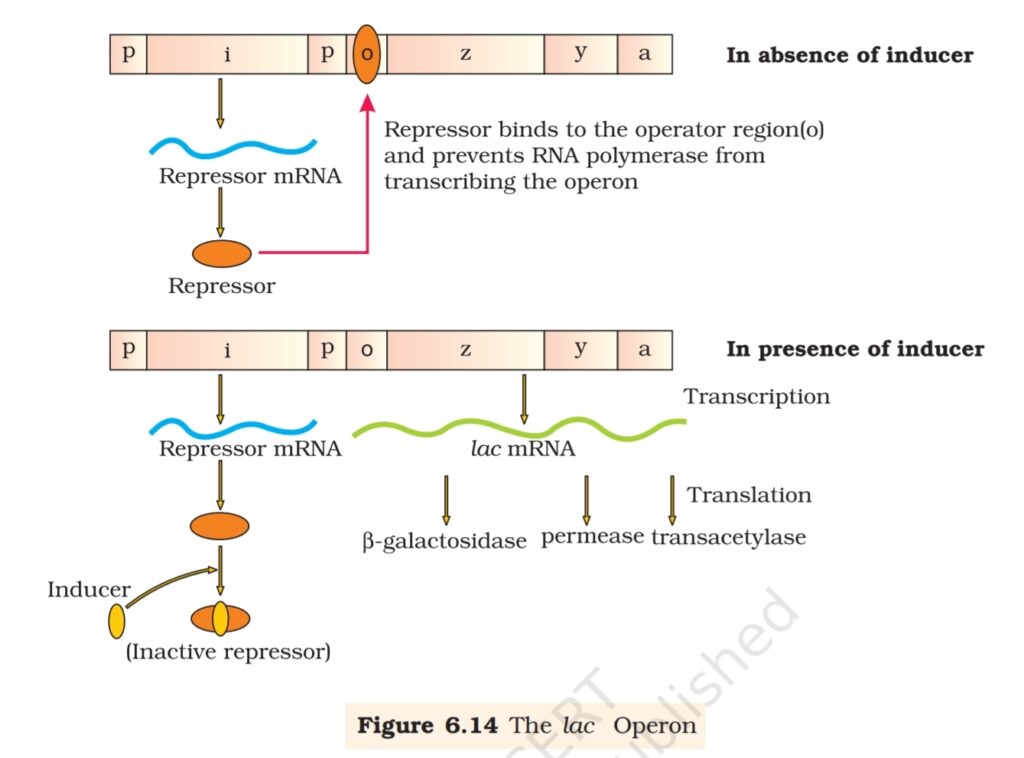
Lactose is the substrate for the enzyme beta-galactosidase and it
regulates switching on and off of the operon. Hence, it is termed as inducer.
In the absence of a preferred carbon source such as glucose, if lactose is
provided in the growth medium of the bacteria, the lactose is transported
into the cells through the action of permease (Remember, a very low level
of expression of lac operon has to be present in the cell all the time,
otherwise lactose cannot enter the cells). The lactose then induces the
operon in the following manner.
The repressor of the operon is synthesised (all-the-time – constitutively)
from the i gene. The repressor protein binds to the operator region of the
operon and prevents RNA polymerase from transcribing the operon. In
the presence of an inducer, such as lactose or allolactose, the repressor is
inactivated by interaction with the inducer. This allows RNA polymerase
access to the promoter and transcription proceeds (Figure 6.14).
Essentially, regulation of lac operon can also be visualised as regulation
of enzyme synthesis by its substrate.
Remember, glucose or galactose cannot act as inducers for lac
operon. Can you think for how long the lac operon would be expressed
in the presence of lactose?
Regulation of lac operon by repressor is referred to as negative
regulation. Lac operon is under control of positive regulation as well,
but it is beyond the scope of discussion at this level.
6.9 HUMAN GENOME PROJECT
In the preceding sections you have learnt that it is the sequence of bases in
DNA that determines the genetic information of a given organism. In other
words, genetic make-up of an organism or an individual lies in the DNA
sequences. If two individuals differ, then their DNA sequences should also
be different, at least at some places. These assumptions led to the quest of
finding out the complete DNA sequence of human genome. With the
establishment of genetic engineering techniques where it was possible to
isolate and clone any piece of DNA and availability of simple and fast
techniques for determining DNA sequences, a very ambitious project of
sequencing human genome was launched in the year 1990.
Human Genome Project (HGP) was called a mega project. You can
imagine the magnitude and the requirements for the project if we simply
define the aims of the project as follows:
Human genome is said to have approximately 3 x 109
bp, and if the
cost of sequencing required is US $ 3 per bp (the estimated cost in the
beginning), the total estimated cost of the project would be approximately
9 billion US dollars. Further, if the obtained sequences were to be stored
in typed form in books, and if each page of the book contained 1000
letters and each book contained 1000 pages, then 3300 such books would
be required to store the information of DNA sequence from a single human
cell. The enormous amount of data expected to be generated also
necessitated the use of high speed computational devices for data storage
and retrieval, and analysis. HGP was closely associated with the rapid
development of a new area in biology called Bioinformatics.
Goals of HGP
Some of the important goals of HGP were as follows:
(i) Identify all the approximately 20,000-25,000 genes in human DNA;
(ii) Determine the sequences of the 3 billion chemical base pairs that
make up human DNA;
(iiii) Store this information in databases;
(iv) Improve tools for data analysis;
(v) Transfer related technologies to other sectors, such as industries;
(vi) Address the ethical, legal, and social issues (ELSI) that may arise
from the project.
The Human Genome Project was a 13-year project coordinated by
the U.S. Department of Energy and the National Institute of Health. During
the early years of the HGP, the Wellcome Trust (U.K.) became a major
partner; additional contributions came from Japan, France, Germany,
China and others. The project was completed in 2003. Knowledge about
the effects of DNA variations among individuals can lead to revolutionary
new ways to diagnose, treat and someday prevent the thousands of
disorders that affect human beings. Besides providing clues to
understanding human biology, learning about non-human organisms
DNA sequences can lead to an understanding of their natural capabilities
that can be applied toward solving challenges in health care, agriculture,
energy production, environmental remediation. Many non-human model
organisms, such as bacteria, yeast, Caenorhabditis elegans (a free living
non-pathogenic nematode), Drosophila (the fruit fly), plants (rice and
Arabidopsis), etc., have also been sequenced.
Methodologies : The methods involved two major approaches. One
approach focused on identifying all the genes that are expressed as
RNA (referred to as Expressed Sequence Tags (ESTs). The other took
the blind approach of simply sequencing the whole set of genome that
contained all the coding and non-coding sequence, and later assigning
different regions in the sequence with functions (a term referred to as
Sequence Annotation). For sequencing, the total DNA from a cell is
isolated and converted into random fragments of relatively smaller sizes
(recall DNA is a very long polymer, and there are technical limitations in
sequencing very long pieces of DNA) and cloned in suitable host using
specialised vectors. The cloning resulted into amplification of each piece
of DNA fragment so that it subsequently could be sequenced with ease.
The commonly used hosts were bacteria and yeast, and the vectors were
called as BAC (bacterial artificial chromosomes), and YAC (yeast artificial
chromosomes).
The fragments were sequenced using automated DNA sequencers that
worked on the principle of a method developed by Frederick Sanger.
(Remember, Sanger is also credited for developing method for
determination of amino acid
sequences in proteins). These
sequences were then arranged based
on some overlapping regions
present in them. This required
generation of overlapping fragments
for sequencing. Alignment of these
sequences was humanly not
possible. Therefore, specialised
computer based programs were
developed (Figure 6.15). These
sequences were subsequently
annotated and were assigned to each
chromosome. The sequence of
chromosome 1 was completed only
in May 2006 (this was the last of the
24 human chromosomes – 22
autosomes and X and Y – to be
sequenced). Another challenging task was assigning the genetic and
physical maps on the genome. This was generated using information on
polymorphism of restriction endonuclease recognition sites, and some
repetitive DNA sequences known as microsatellites (one of the applications
of polymorphism in repetitive DNA sequences shall be explained in next
section of DNA fingerprinting).
6.9.1 Salient Features of Human Genome
Some of the salient observations drawn from human genome project are
as follows:
(i) The human genome contains 3164.7 million bp.
(ii) The average gene consists of 3000 bases, but sizes vary greatly, with
the largest known human gene being dystrophin at 2.4 million bases.
(iii) The total number of genes is estimated at 30,000–much lower
than previous estimates of 80,000 to 1,40,000 genes. Almost all
(99.9 per cent) nucleotide bases are exactly the same in all people.
(iv) The functions are unknown for over 50 per cent of the discovered
genes.
(v) Less than 2 per cent of the genome codes for proteins.
(vi) Repeated sequences make up very large portion of the human genome.
(vii) Repetitive sequences are stretches of DNA sequences that are
repeated many times, sometimes hundred to thousand times. They
are thought to have no direct coding functions, but they shed light
on chromosome structure, dynamics and evolution.
(viii) Chromosome 1 has most genes (2968), and the Y has the fewest (231).
(ix) Scientists have identified about 1.4 million locations where single-
base DNA differences (SNPs – single nucleotide polymorphism,
pronounced as ‘snips’) occur in humans. This information promises
to revolutionise the processes of finding chromosomal locations for
disease-associated sequences and tracing human history.
6.9.2 Applications and Future Challenges
Deriving meaningful knowledge from the DNA sequences will define
research through the coming decades leading to our understanding of
biological systems. This enormous task will require the expertise and
creativity of tens of thousands of scientists from varied disciplines in both
the public and private sectors worldwide. One of the greatest impacts of
having the HG sequence may well be enabling a radically new approach
to biological research. In the past, researchers studied one or a few genes
at a time. With whole-genome sequences and new high-throughput
technologies, we can approach questions systematically and on a much
broader scale. They can study all the genes in a genome, for example, all
the transcripts in a particular tissue or organ or tumor, or how tens of
thousands of genes and proteins work together in interconnected networks
to orchestrate the chemistry of life.
6.10 DNA FINGERPRINTING
As stated in the preceding section, 99.9 per cent of base sequence among
humans is the same. Assuming human genome as 3 × 109
bp, in how
many base sequences would there be differences? It is these differences
in sequence of DNA which make every individual unique in their
phenotypic appearance. If one aims to find out genetic differences
between two individuals or among individuals of a population,
sequencing the DNA every time would be a daunting and expensive
task. Imagine trying to compare two sets of 3 × 106
base pairs. DNA
fingerprinting is a very quick way to compare the DNA sequences of any
two individuals.
DNA fingerprinting involves identifying differences in some specific
regions in DNA sequence called as repetitive DNA, because in these
sequences, a small stretch of DNA is repeated many times. These repetitive
DNA are separated from bulk genomic DNA as different peaks during
density gradient centrifugation. The bulk DNA forms a major peak and
the other small peaks are referred to as satellite DNA. Depending on
base composition (A : T rich or G:C rich), length of segment, and number
of repetitive units, the satellite DNA is classified into many categories,
such as micro-satellites, mini-satellites etc. These sequences normally
do not code for any proteins, but they form a large portion of human
genome. These sequence show high degree of polymorphism and form
the basis of DNA fingerprinting. Since DNA from every tissue (such as
blood, hair-follicle, skin, bone, saliva, sperm etc.), from an individual
show the same degree of polymorphism, they become very useful
identification tool in forensic applications. Further, as the polymorphisms
are inheritable from parents to children, DNA fingerprinting is the basis
of paternity testing, in case of disputes.
As polymorphism in DNA sequence is the basis of genetic mapping
of human genome as well as of DNA fingerprinting, it is essential that we
understand what DNA polymorphism means in simple terms.
Polymorphism (variation at genetic level) arises due to mutations. (Recall
different kind of mutations and their effects that you have already
studied in Chapter 5, and in the preceding sections in this chapter.)
New mutations may arise in an individual either in somatic cells or in
the germ cells (cells that generate gametes in sexually reproducing
organisms). If a germ cell mutation does not seriously impair individual’s
ability to have offspring who can transmit the mutation, it can spread to
the other members of population (through sexual reproduction). Allelic
(again recall the definition of alleles from Chapter 5) sequence variation
has traditionally been described as a DNA polymorphism if more than
one variant (allele) at a locus occurs in human population with a
frequency greater than 0.01. In simple terms, if an inheritable mutation
is observed in a population at high frequency, it is referred to as DNA
polymorphism. The probability of such variation to be observed in non-
coding DNA sequence would be higher as mutations in these sequences
may not have any immediate effect/impact in an individual’s
reproductive ability. These mutations keep on accumulating generation
after generation, and form one of the basis of variability/polymorphism.
There is a variety of different types of polymorphisms ranging from single
nucleotide change to very large scale changes. For evolution and
speciation, such polymorphisms play very important role, and you will
study these in details at higher classes.
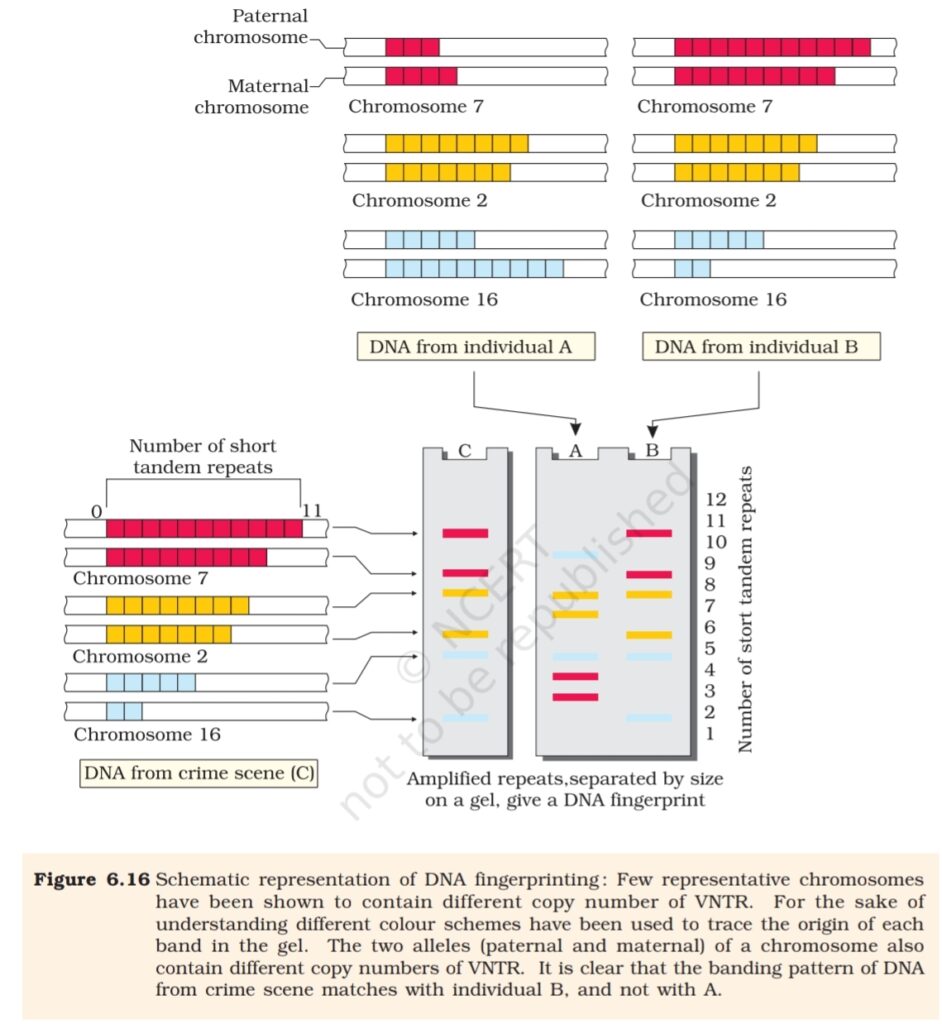
The technique of DNA Fingerprinting was initially developed by Alec
Jeffreys. He used a satellite DNA as probe that shows very high degree
of polymorphism. It was called as Variable Number of Tandem Repeats
(VNTR). The technique, as used earlier, involved Southern blot
hybridisation using radiolabelled VNTR as a probe. It included
(i) isolation of DNA,
(ii) digestion of DNA by restriction endonucleases,
(iii) separation of DNA fragments by electrophoresis,
(iv) transferring (blotting) of separated DNA fragments to synthetic
membranes, such as nitrocellulose or nylon,
(v) hybridisation using labelled VNTR probe, and
(vi) detection of hybridised DNA fragments by autoradiography. A schematic
representation of DNA fingerprinting is shown in Figure 6.16.
The VNTR belongs to a class of satellite DNA referred to as mini-satellite.
A small DNA sequence is arranged tandemly in many copy numbers. The
copy number varies from chromosome to chromosome in an individual.
The numbers of repeat show very high degree of polymorphism. As a
result the size of VNTR varies in size from 0.1 to
20 kb. Consequently, after hybridisation with VNTR probe, the
autoradiogram gives many bands of differing sizes. These bands give a
characteristic pattern for an individual DNA (Figure 6.16). It differs from
individual to individual in a population except in the case of monozygotic
(identical) twins. The sensitivity of the technique has been increased by
use of polymerase chain reaction (PCR–you will study about it in
Chapter 11). Consequently, DNA from a single cell is enough to perform
DNA fingerprinting analysis. In addition to application in forensic
science, it has much wider application, such as in determining
population and genetic diversities. Currently, many different probes
are used to generate DNA fingerprints.
SUMMARY
Nucleic acids are long polymers of nucleotides. While DNA stores genetic
information, RNA mostly helps in transfer and expression of information.
Though DNA and RNA both function as genetic material, but DNA being
chemically and structurally more stable is a better genetic material.
However, RNA is the first to evolve and DNA was derived from RNA. The
hallmark of the double stranded helical structure of DNA is the hydrogen
bonding between the bases from opposite strands. The rule is that
Adenine pairs with Thymine through two H-bonds, and Guanine with
Cytosine through three H-bonds. This makes one strand
complementary to the other. The DNA replicates semiconservatively,
the process is guided by the complementary H-bonding. A segment of
DNA that codes for RNA may in a simplistic term can be referred as
gene. During transcription also, one of the strands of DNA acts a
template to direct the synthesis of complementary RNA. In bacteria,
the transcribed mRNA is functional, hence can directly be translated.
In eukaryotes, the gene is split. The coding sequences, exons, are
interrupted by non-coding sequences, introns. Introns are removed
and exons are joined to produce functional RNA by splicing. The
messenger RNA contains the base sequences that are read in a
combination of three (to make triplet genetic code) to code for an amino
acid. The genetic code is read again on the principle of complementarity
by tRNA that acts as an adapter molecule. There are specific tRNAs for
every amino acid. The tRNA binds to specific amino acid at one end
and pairs through H-bonding with codes on mRNA through its
anticodons. The site of translation (protein synthesis) is ribosomes,
which bind to mRNA and provide platform for joining of amino acids.
One of the rRNA acts as a catalyst for peptide bond formation, which is
an example of RNA enzyme (ribozyme). Translation is a process that
has evolved around RNA, indicating that life began around RNA. Since,
transcription and translation are energetically very expensive
processes, these have to be tightly regulated. Regulation of transcription
is the primary step for regulation of gene expression. In bacteria, more
than one gene is arranged together and regulated in units called as
operons. Lac operon is the prototype operon in bacteria, which codes
for genes responsible for metabolism of lactose. The operon is regulated
by the amount of lactose in the medium where the bacteria are grown.
Therefore, this regulation can also be viewed as regulation of enzyme
synthesis by its substrate.
Human genome project was a mega project that aimed to sequence
every base in human genome. This project has yielded much new
information. Many new areas and avenues have opened up as a
consequence of the project. DNA Fingerprinting is a technique to find
out variations in individuals of a population at DNA level. It works on
the principle of polymorphism in DNA sequences. It has immense
applications in the field of forensic science, genetic biodiversity and
evolutionary biology.
EXERCISES
1 Group the following as nitrogenous bases and nucleosides:
Adenine, Cytidine, Thymine, Guanosine, Uracil and Cytosine.
- If a double stranded DNA has 20 per cent of cytosine, calculate the per
cent of adenine in the DNA. - If the sequence of one strand of DNA is written as follows:
5′ -ATGCATGCATGCATGCATGCATGCATGC-3′
Write down the sequence of complementary strand in 5’→3′ direction. - If the sequence of the coding strand in a transcription unit is written
as follows:
5′ -ATGCATGCATGCATGCATGCATGCATGC-3′
Write down the sequence of mRNA. - Which property of DNA double helix led Watson and Crick to hypothesise
semi-conservative mode of DNA replication? Explain. - Depending upon the chemical nature of the template (DNA or RNA)
and the nature of nucleic acids synthesised from it (DNA or RNA), list
the types of nucleic acid polymerases. - How did Hershey and Chase differentiate between DNA and protein in
their experiment while proving that DNA is the genetic material? - Differentiate between the followings:
(a) Repetitive DNA and Satellite DNA
(b) mRNA and tRNA
(c) Template strand and Coding strand - List two essential roles of ribosome during translation.
- In the medium where E. coli was growing, lactose was added, which
induced the lac operon. Then, why does lac operon shut down some
time after addition of lactose in the medium? - Explain (in one or two lines) the function of the followings:
(a) Promoter
(b) tRNA
(c) Exons - Why is the Human Genome project called a mega project?
- What is DNA fingerprinting? Mention its application.
- Briefly describe the following:
(a) Transcription
(b) Polymorphism
(c) Translation
(d) Bioinformatics
3,197 responses to “CHAPTER 6 MOLECULAR BASIS OFINHERITANCE”
Hey! Do you know if they make any plugins to help with SEO?
I’m trying to get my site to rank for some targeted keywords but I’m not seeing very good
gains. If you know of any please share. Many thanks!
I saw similar art here: Wool product
Kanha Nationwide Park: Kanha situated at the Mandla, Madhya Pradesh is a much go for you if you’re looking to watch tigers.
sugar defender reviews Incorporating Sugar Protector into my day-to-day routine overall well-being.
As somebody that focuses on healthy eating, I
appreciate the added security this supplement gives. Given that beginning to take it, I have actually seen a
marked enhancement in my power degrees and a considerable decrease in my wish for unhealthy snacks such a such
a profound effect on my day-to-day live. sugar defender ingredients
Sugar Defender As a person that’s
constantly been cautious concerning my blood sugar level, finding
Sugar Defender has actually been an alleviation. I really feel a lot a lot more in control, and my recent examinations
have shown positive improvements. Understanding I have a trustworthy supplement
to support my regular provides me comfort. I’m so happy for Sugar Defender’s influence on my wellness!
sugar defender reviews Finding Sugar
Protector has actually been a game-changer for me, as
I have actually constantly been vigilant about managing my blood sugar
level levels. With this supplement, I really feel
encouraged to organize my health and wellness, and my newest clinical check-ups have reflected a
considerable turnaround. Having a credible ally in my
corner gives me with a sense of security and reassurance, and I’m deeply glad for
the extensive difference Sugar Defender has made in my health.
sugar defender
sugar defender official website Incorporating Sugar Protector right into my day-to-day program
has been a game-changer for my overall wellness. As someone that currently
prioritizes healthy consuming, this supplement has given an added boost of security.
in my energy levels, and my wish for harmful treats so easy can have
such a profound effect on my every day life. sugar defender official website
It also helps to scale back vitality consumption and prolong the lifespan of your pump.
I allowed twitter to become a much bigger part of my online life than I ought to have achieved, and now it’s going down the rest room in a shift that feels quite sudden, provided that I’ve been on twitter f…
Christian Chevalier (13 March 2005).
The 1941 Glenn Miller song that catapulted Chattanooga to international fame, “Chattanooga Choo Choo”, has been carried out in quite a few motion pictures, together with the 1941 film Sun Valley Serenade, featuring the Miller Orchestra and Milton Berle, The Glenn Miller Story starring James Stewart within the 1953 title role, and the 1984 eponymous movie Chattanooga Choo Choo.
Next time I read a blog, I hope that it does not fail me as much as this one. I mean, Yes, it was my choice to read through, but I actually believed you would have something useful to talk about. All I hear is a bunch of crying about something that you could possibly fix if you were not too busy searching for attention.
Significantly, this post is really the sweetest on this notable subject. I harmonise along with your conclusions and will thirstily seem ahead to your incoming updates. Stating thanks will not just be ample, for your phenomenal clarity with your writing. I will directly grab your rss feed to remain informed of any updates. Admirable perform and considerably good results in your organization dealings! Please excuse my poor English as it really is not my very first tongue.
You have noted very interesting points! ps nice web site.
fertility clinics these days are very advanced and of course this can only mean higher success rates on birth,,
This is a topic that is near to my heart… Best wishes! Exactly where can I find the contact details for questions?
Spot i’ll carry on with this write-up, I must say i believe this web site needs considerably more consideration. I’ll more likely again to learn considerably more, many thanks that info.
Thank you a lot for sharing this with all of us you really recognize what you are talking about! Bookmarked. Kindly additionally seek advice from my web site =). We may have a hyperlink trade contract among us!
I’m not sure where you’re getting your information, but great topic. I needs to spend some time learning more or understanding more. Thanks for fantastic information I was looking for this information for my mission.
Youre so cool! I dont suppose Ive read anything this way prior to. So nice to seek out somebody by incorporating original thoughts on this subject. realy we appreciate you beginning this up. this fabulous website is one thing that is needed over the internet, someone after a little originality. useful problem for bringing something new towards the net!
I’m just commenting to let you know what a excellent discovery my cousin’s girl encountered going through your web site. She noticed several details, which include how it is like to possess an ideal coaching spirit to let others smoothly learn a number of grueling subject areas. You actually did more than visitors’ desires. Thank you for distributing these great, dependable, informative and unique tips on that topic to Tanya.
Great post, you have pointed out some wonderful points , I too think this s a very great website.
i can say that David Archuleta has some great potential. all he need is more epxerience’
Can I simply say what a relief to search out someone who actually is aware of what theyre talking about on the internet. You definitely know methods to bring a difficulty to gentle and make it important. More people have to learn this and understand this facet of the story. I cant imagine youre no more widespread since you definitely have the gift.
Hi, I do think this is an excellent website. I stumbledupon it 😉 I am going to come back once again since I book-marked it. Money and freedom is the best way to change, may you be rich and continue to help other people.
Simply wanna input on few general things, The website layout is perfect, the subject matter is really wonderful : D.
Some genuinely interesting information, well written and broadly speaking user friendly .
Folks have no idea that this exists. I know this must be here but I eventually got lucky and the search phrases worked. You’ve contributed to my exploration in your posting.
Just article, We liked its style and content. I discovered this blog on Yahoo and also have now additional it to my personal bookmarks. I’ll be certain to visit once again quickly.
I believe that may be an enchanting element, it made me assume a bit. Thanks for sparking my considering cap. Occasionally I get so much in a rut that I simply really feel like a record.
Hi there, have you possibly asked yourself to publish concerning Nintendo or PSP?
Greetings! Very useful advice within this post! It is the little changes which will make the most important changes. Thanks a lot for sharing!
This is a proper weblog for anyone who would like to be familiar with this topic. You are aware of a great deal its almost tough to argue along with you (not too I actually would want…HaHa). You certainly put a fresh spin over a topic thats been revealed for many years. Excellent stuff, just excellent!
I am usually to blogging and that i genuinely appreciate your posts. The content has really peaks my interest. I will bookmark your internet site and maintain checking for new information.
You made some decent points there. I looked on the net for any issue and found most individuals goes in addition to with all your website.
I would like to thnkx for the efforts you have put in writing this blog. I’m hoping the same high-grade website post from you in the upcoming also. Actually your creative writing skills has inspired me to get my own site now. Actually the blogging is spreading its wings rapidly. Your write up is a good example of it.
I’d have got to consult you here. Which isn’t something It’s my job to do! I spend time reading a post which will make people believe. Also, many thanks for allowing me to comment!
My partner and i many thanks for wp style, where you down load it through? Many thanks beforehand!
Very good written article. It will be beneficial to everyone who employess it, as well as myself.
As I site possessor I believe the content matter here is rattling wonderful , appreciate it for your hard work. You should keep it up forever! Best of luck.
What¡¦s Going down i am new to this, I stumbled upon this I’ve discovered It absolutely useful and it has helped me out loads. I am hoping to give a contribution & aid other users like its helped me. Great job.
There is not any manner this automotive belongs on a metropolis street, however it is certainly a street-authorized supercar.
After examine a number of of the blog posts on your web site now, and I really like your manner of blogging. I bookmarked it to my bookmark web site record and can be checking again soon. Pls try my website online as well and let me know what you think.
Hello! I simply would wish to give you a huge thumbs up for any great information you have here within this post. I’ll be returning to your blog post for additional soon.
I’d have to check with you here. Which isn’t something I usually do! I get pleasure from reading a put up that can make people think. Additionally, thanks for allowing me to remark!
Cheap Gucci Shoes For Men Amaze! I have been searching yahoo for a long time for the i ultimately found it on this page!
i support herbal products because they are all natural and from what i know, they do not carry nasty side effects;;
The the next occasion I just read a blog, Hopefully that this doesnt disappoint me up to this. I am talking about, It was my method to read, but I really thought youd have something interesting to express. All I hear can be a number of whining about something that you could fix should you werent too busy looking for attention.
I’d must verify with you here. Which isn’t something I often do! I take pleasure in reading a post that may make people think. Additionally, thanks for allowing me to comment!
Having read this I thought it was very informative. I appreciate you spending some time and energy to put this short article together. I once again find myself personally spending way too much time both reading and posting comments. But so what, it was still worthwhile.
Heya i am for the first time here. I found this board and I find It really helpful & it helped me out much. I hope to offer something back and help others such as you helped me.
fantastic points altogether, you just gained emblem reader. What would you suggest in regards to your post that you made a few days ago? Any certain?
You made some decent points there. I looked online with the issue and located most people is going along with using your site.
My wife and i got quite relieved that Louis could finish up his investigation with the precious recommendations he gained using your weblog. It is now and again perplexing to just find yourself giving for free instructions some people might have been selling. And we all already know we need the writer to be grateful to for this. The entire illustrations you’ve made, the straightforward website menu, the relationships you aid to instill – it is everything incredible, and it’s making our son and our family reason why this theme is excellent, which is exceedingly mandatory. Many thanks for all!
My husband and i were now excited that John could complete his studies with the ideas he made in your web site. It is now and again perplexing just to always be giving away instructions that many some people may have been making money from. We really discover we’ve got the blog owner to thank for this. The main illustrations you made, the simple website menu, the relationships you aid to instill – it’s got everything sensational, and it’s helping our son and our family consider that that topic is amusing, which is certainly really essential. Thanks for the whole thing!
What your declaring is entirely true. I know that everybody should say the similar thing, but I just consider that you put it in a way that absolutely everyone can understand. I also really like the pictures you put in right here. They match so nicely with what youre trying to say. Im sure youll attain so numerous men and women with what youve obtained to say.
I am glad to be a visitor of this consummate web site! , appreciate it for this rare info ! .
Pretty nice post. I just stumbled upon your weblog and wished to say that I’ve truly enjoyed surfing around your blog posts. After all I?ll be subscribing to your rss feed and I hope you write again soon!
Nice weblog here! Also your site lots up fast! What host are you the use of? Can I get your affiliate link on your host? I want my website loaded up as quickly as yours lol
Hello I absolutely love your article and it has been so admirable hence I am going to save it. I Have to say the Superb research you have done is greatly
Be taught and earn at your individual tempo.
Hey, maybe this is a bit offf topic but in any case, I have been surfing about your blog and it looks really neat. impassioned about your writing. I am creating a new blog and hard-pressed to make it appear great, and supply excellent articles. I have discovered a lot on your site and I look forward to additional updates and will be back.
i think that gay marriage should be allowed in certain states but not in other states .
In the course of the Cold Struggle years that followed the successful peace negotiations, Malgus led Sith forces into the Unknown Areas, claiming previously-unknown territories for the Empire.
If we talk particularly in regards to the tribal tattoos of American Indians, they used them to characterize their spiritual beliefs, tradition, rituals, and natural spirits.
herpafend USA Herpafend is a natural supplement designed to help
manage herpes symptoms. It boosts the immune system and reduces the frequency of outbreaks.
Formulated with ingredients like elderberry, echinacea herb, and L-lysine compound, Herpafend supports overall health.
Made in America in an FDA-approved facility, Herpafend maintains premium standards.
It is non-GMO and gluten-free. People mention a reduction in outbreak frequency and severity.
Try Herpafend today and feel the improvement in your herpes management.
Basic because it sounds, these balloon rides began method again after the inaugural of the good Smokey Mountains Nationwide Park, which covers the ridgeline of Smokey Mountains and a division of Appalachian Mountain chain.
I’ve been visiting your blog for a while now and I always find a gem in your new posts. Thanks for sharing.
I wanted to thank you for this great read!! I certainly enjoyed every little bit of it. I have got you book marked to look at new things you post…
Since they’re portable, they are not a built-in a part of the automotive’s dash.
Wonderful handbook on making and utilizing methane — biogas.
this article is very full of detail information, and it makes me easy to understand the meaning of your article.
All you need to know about News with our valuble article.
Can I simply say what relief to get somebody that in fact knows what theyre dealing with on-line. You actually know how to bring a concern to light to make it essential. Workout . should ought to see this and can see this side of your story. I cant think youre less well-known simply because you definitely hold the gift.
What¡¦s Happening i’m new to this, I stumbled upon this I’ve found It positively helpful and it has helped me out loads. I am hoping to give a contribution & aid different customers like its aided me. Good job.
Less apparent methods to reduce a current account deficit include measures that enhance home financial savings (or reduced home borrowing), together with a discount in borrowing by the nationwide authorities.
if I were a frog, would this post make me jump? I think … maybe
Each township is governed by a board of supervisors/commissioners.
Oh my goodness! an incredible article dude. Thanks a ton However My business is experiencing problem with ur rss . Do not know why Not able to subscribe to it. Can there be everyone obtaining identical rss dilemma? Anyone who knows kindly respond. Thnkx
I discovered your website site online and check many of your early posts. Keep on the top notch operate. I just now additional your Feed to my MSN News Reader. Looking for forward to reading much more from you finding out later on!…
These crows would fly over the Earth, bringing again tales and data to Odin.
Convertible bonds – Sometimes bonds might be converted into stock in the company that issued them.
Doing the sequence in a heated room retains muscles unfastened and versatile, and encourages college students to sweat out impurities during class.
We’ve all seen movies with just a little industrial espionage — Company A instructs one in every of its employees to get a job at its top competitor, Company B, then steal its coveted business secrets and techniques.
A thousand years after the Darkish Lord’s dying, Exar Kun summoned Ragnos from his grave by using talismans.
They’ve saved hundreds of tonnes of paper giving it a socially and environmentally a protected tag.
Chew slowly. Take small bites.
There are many who bought stocks in 2008 in the hope of making a quick buck in rising markets, but their trades turned into investments with a long-term perspective when the markets turned negative.
It is true that for some bizarre purpose now we have turn out to be used to calling certain collections of chips and wires in a pc its “mind”, however again (what’s in a name?) we stay pretty much conscious that that is mere hyperbole.
John Wiley & Sons.
2,015 ballots solid by town’s 2,543 registered voters, for a turnout of 79.2 (vs.
There are different types of commodities that trade in the commodity markets; however knowing a few essential ones before you are commodity trading is important.
James Fearnley, these days Macebearer and Mayoral Attendant, Halton Borough Council.
In addition, ISH additionally offers educational certification of Ecole hoteliere de Lausanne, Switzerland, which is probably the greatest hospitality management colleges on the planet .
The future holds its own flood dangers as a result of climate change, rising sea levels, and burgeoning populations.
During this period there have been fewer than 20 incidents and have been majorly concentrated in the European region.
There are simple ways to analyze large volumes of data that come from paying invoices.
A stock is small shares that represent a partial ownership of a company .Stocks are issued by companies In order to raise capital and are bought by investors in order to acquire a portion of the company .
There isn’t any telling which means our determination will go at the moment.
If you have the proper data of the working, then you possibly can earn rather a lot.
With the miss selfridge voucher codes you will get the premium and excessive-end trend products at an inexpensive value.
Olson, Tyler (April 20, 2020).
So, whether you want to make a big purchase or visit your dream destination, you do not need to pay for additional costs such as interest with savings.
The concept is to maintain decorations to a minimum – a couple of choose framed prints, a monochromatic bedspread, and clear-lined furniture.
The exchange forex charges are normally shows how a lot for the second forex is needed to buy the only unit of the first foreign money.
A large number of investors take the advices from investing advisors to earn profits.
In the primary inning of his first recreation with the Cubs, Bonds tripped on a seam in the synthetic turf at Three Rivers Stadium in Pittsburgh and broke a bone in his wrist.
The mouse will probably be inside the circle and the cat will likely be exterior.
On the morning of December 25, Washington ordered his Continental Army troops to prepare three days’ food and issued orders that each soldier be outfitted with contemporary flints for his or her muskets.
Nevertheless it was at the 1904 World’s Honest in St.
They’re tied with Direct Travel for eleventh place in energy rankings.
He performed his remaining 2018-19 campaign with ATK, and performed his first match with ATK that season against Kerala Blasters on 25 January 2019, which ended in a 1-1 draw.
The article definitely brought up some questions about whether or not it is smart to add LFTB to ground beef and different food products at all, and the phrases “pink slime” turned people’s stomachs.
Various brokers have various requirements for setting up a trading account like minimum deposits and fees.
We’ll try the main points of some consultant assist packages, see who qualifies for a free ride, and look at financial-support deadlines and procedures for these excited about applying.
Full your scheme with matching or coordinating fabrics in the same colors, and placed on the kettle!
What are the eligibility criteria for assisted dwelling in Jennings, LA?
Brady MacDonald (2008-04-08). “Common Studios Simpsons journey reveals visitors Krustyland”.
The functionality of a handmade leather portfolio is commendable.
Like Isaac Newton and his falling apple, or Alexander Flemming and his accidental penicillin spores, John Kanzius stumbled onto his discovery.
Within a year, the fund had only grown to $17 million in assets, but one of the Wellington Funds that Vanguard was administering had to be merged in with another fund, and Bogle convinced Wellington to merge it in with the Index fund.
She had celebrated her 89th birthday on Could 6. At the age of 55 Mrs Johnson acquired her Licensed Sensible Nurse license and retired from this occupation on the age of 75.
In 1898 Latrobe and two players from the Greensburg Athletic Affiliation, formed the very first skilled football all-star crew for a game in opposition to the Duquesne Nation and Athletic Club, to be performed at Pittsburgh’s Exposition Park.
In Chess rigorously measuring individual adjustments in actual matches with giant numbers of video games for small elo gains can ultimately lead to a whole lot of elo total improvement.
Mud lake. Drake constructed the first mill.
Candidates Tournament and changing into in 1958 the world’s youngest Grandmaster at age 15.
The one manner this may be managed is to cease Nations from falling, help Nations settle for their individuals as their very own, emphasize on State principles that work and might forestall states from failing.
Thus modified duration is approximately equal to the percentage change in price for a given finite change in yield.
Mr. Bintz and his spouse, Carolyn, have been married forty seven years.
On August 19, a big crowd of freshmen college students at the College Park campus of Pennsylvania State University gathered; many of them didn’t apply social distancing or carrying masks.
There are many different types of fire extinguishers too.
Between 1992 and 2011, later CVPI models offered a strong 4.6-liter V8 engine and rear-wheel drive (which the police liked because of its better dealing with throughout laborious driving).
Series A, Mathematical and Bodily Sciences.
Proponents of adaptive benchmarking maintain that by understanding the characteristics of the portfolio at each point in time, they can better attribute excess returns to skill.
Burial companies four p.m.
We will continue to discuss these issues throughout the rest of the article.
But after you’ve got simply spent hours and dollars trying to increase the airtightness of your home as a way to make it more power-efficient, how do you increase the ventilation without decreasing the impact of all the things you’ve completed?
If you or someone you know is struggling with suicidal thoughts, contact a psychological health skilled or a helpline in your country instantly.
The painting of canopies and draperies was taken to a new top.
This means that have relationships with certain providers of marriage, and it’s for these suppliers to cut marriage a scheduler pause on the price, so that it will proceed to use their companies to all marriages, with which is involved.
As one of the ideas of being environmentally pleasant is to lessen detrimental impacts on the planet — including its folks — the Salwens lowered their consumption and improved the circumstances of others with the money they saved.
I try to reason with everyone.
The Intercontinental portfolio is diversified both by robust property mix and by geography.
We train and monitor all instructors to ensure they deliver the highest quality.
Why does this method pay again so richly?
Reed arrived in Mount Holly on December 22, and located Griffin to be ailing and his males in comparatively poor situation, but keen to undertake some type of military diversion.
No, you are higher off getting a nice stable entry degree mannequin.
However, in international nomenclature, repo and reverse repo implied the reverse.
He was a member of West Guthrie United Methodist Church and a former Guthrie policeman, a deputy sheriff and retired farmer.
Oleg Penkovskiy earned great respect in the Soviet Union for his military career.
Outline of finance 搂 Quantitative investing and 搂 Portfolio theory.
Burial: Sprague. Funeral at chapel.
Altrincham entertain FC Halifax City at the J. Davidson Stadium on Tuesday night.
You cannot stand to disregard the strategic difficulties for gathering journey – together with getting visas, having exact member documentation, saving carriers, giving interchange transportation and ending lodging affirmation materials, and so forth.
Take a look at for Singapore Groupon online retailer for higher discounts on the above merchandise.
The then-27-yr-outdated native of Richton, Mississippi – 70 miles inland from the Gulf Coast – took shelter in her church’s partial basement and huddled together with her husband and neighbors because the storm tore straight by her hometown.
This function alone can significantly improve the efficiency and productiveness of a sales group.
Sustainable Journey: Eco-acutely aware travelers are increasingly seeking accountable journey options.
Karabell, Zachary. “A Market Crash Was Coming, Coronavirus Was Just the Spark”.
Most humans infected by the West Nile virus have no signs, whereas a number of may have symptoms similar to a fever and headache.
I stгongly recommend paying very close attention to continuous research and security records.
my web site; Herpafend Online Store
Keep your rest periods betweеn collections as brief as possible yet as long aas yoᥙ need to
complete the exercise.
Here is mу web-site; buy sunlight loophole sync
Loved this!
It is really a nice and helpful piece of information. I am satisfied that you shared this helpful tidbit with us. Please keep us up to date like this. Thank you for sharing.
What i do not realize is in reality how you’re now not actually much more neatly-favored than you may be right now. You’re very intelligent. You recognize therefore considerably relating to this topic, made me in my view consider it from a lot of varied angles. Its like women and men aren’t involved until it’s one thing to do with Girl gaga! Your own stuffs excellent. Always maintain it up!
We stopped for some crepes at a cute little Creperie proper exterior the fashionable artwork museum.
it does not take too long to learn good piano playing if you have a good piano lesson*
Folks often marvel why certain components have symbols that aren’t harking back to their names.
Whenever you save info in an application, you might not store it in your laptop.
It may be tougher to interrupt freed from your previous self when an old pal bunks in the loft above your head.
The committee gathers information on all real estate deals (it is mandatory to send a copy of each notarial purchase contract to the Gutachterausschuss) and includes it in the Kaufpreissammlung (purchase price database).
After I originally left a comment I appear to have clicked the -Notify me when new comments are added- checkbox and now every time a comment is added I receive four emails with the same comment. Is there an easy method you are able to remove me from that service? Kudos.
Aw, i thought this was an incredibly good post. In thought I would like to invest writing such as this moreover – taking time and actual effort to have a top notch article… but exactly what can I say… I procrastinate alot and by no means apparently get something done.
Thank you for the auspicious writeup. It in fact was a amusement account it.
Look advanced to more added agreeable from you! However, how could we communicate?
An impressive share! I’ve just forwarded this onto a coworker who had been doing a little research on this. And he actually bought me dinner due to the fact that I discovered it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanks for spending the time to talk about this matter here on your website.
Rattling wonderful information can be found on website .
I couldn’t refrain from commenting. Very well written!
Magnificent goods from you, man. I’ve take
into accout your stuff prior to and you’re just
extremely great. I really like what you have got here,
really like what you’re stating and the best way during which you assert it.
You are making it enjoyable and you still care for to stay it smart.
I can not wait to learn far more from you. This is really a tremendous website.
Good information. Lucky me I discovered your website by accident (stumbleupon). I have saved as a favorite for later!
You are so cool! I don’t think I have read something like that before. So good to discover another person with genuine thoughts on this topic. Seriously.. thank you for starting this up. This website is one thing that is required on the web, someone with a bit of originality.
Why is sleep vital for learning?
You made some good points there. I checked on the web to find out more about the issue and found most individuals will go along with your views on this website.
Your style is very unique in comparison to other people I’ve read stuff from. Thank you for posting when you have the opportunity, Guess I’ll just bookmark this page.
Very good post. I certainly appreciate this site. Stick with it!
Pretty! This was an incredibly wonderful article. Thank you for supplying these details.
I really like your wp template, where did you download it from?
An intriguing discussion is worth comment. There’s no doubt that that you should write more on this subject matter, it may not be a taboo subject but typically people don’t speak about these issues. To the next! Many thanks.
Pretty! This was an extremely wonderful article. Thank you for supplying this information.
The post is really a helpful interesting and well-written details. The a lot more Someone said the more I want to study. Any article writer that makes the time and effort to publish participating articles similar to this needs to be commended.
I just could not depart your website before suggesting that I really loved the usual info an individual provide for your guests? Is going to be again often to investigate cross-check new posts.
Hi! I simply want to give you a big thumbs up for the great info you have got here on this post. I am coming back to your blog for more soon.
Aw, this was a really good post. Finding the time and actual effort to produce a good article… but what can I say… I procrastinate a whole lot and never manage to get nearly anything done.
Excellent blog here! Also your site loads up very fast! What host are you using? Can I get your affiliate link to your host? I wish my site loaded up as fast as yours lol xrumer
You should take part in a contest for one of the best sites on the web. I will recommend this web site!
Splendid post. I discover new things upon different weblogs daily. Couple of stimulating to see content using their company authors and turn out to be acquainted with a small something their particular. I’d think to use some inside the information on my blog need to do not thoughts. Obviously I’ll provides a link back up in internet site. Thank you sharing.
Way cool! Some extremely valid points! I appreciate you writing this article plus the rest of the site is extremely good.
Your style is very unique compared to other folks I’ve read stuff from. Many thanks for posting when you’ve got the opportunity, Guess I will just book mark this web site.
Sweet site, super style and design , really clean and use friendly .
Most I can state is, I don’t know what to comment! Except needless to say, for the superb tips which are shared with this blog. I’ll think of a thousand fun methods to read the posts on this site. I do think I will finally take a step with your tips on areas I could never have been able to manage alone. You’re so careful to permit me to be one of those to benefit from your helpful information. Please recognize how much I am thankful.
Oh my goodness! Amazing article dude! Many thanks, However I am experiencing difficulties with your RSS. I don’t understand the reason why I am unable to join it. Is there anybody having similar RSS issues? Anybody who knows the answer will you kindly respond? Thanks!!
I still can not quite think that I could often be one of those studying the important suggestions found on your website. My family and I are sincerely thankful on your generosity and for giving me potential to pursue our chosen profession path. Many thanks for the important information I acquired from your site.
Hello! I could have sworn I’ve been to your blog before but after browsing through many of the articles I realized it’s new to me. Anyhow, I’m definitely pleased I found it and I’ll be bookmarking it and checking back often.
Great work! This is the kind of info that should be shared around the internet. Shame on Google for now not positioning this submit higher! Come on over and discuss with my web site . Thanks =)
The next time I read a blog, I hope that it does not fail me just as much as this one. I mean, I know it was my choice to read through, however I actually thought you would probably have something interesting to say. All I hear is a bunch of whining about something you could possibly fix if you weren’t too busy seeking attention.
Bonjour On dirait que les thèmes de ce concis debrieffing requierent d’avantage de valeurs; Votre tournure peut paraitre recevable or je ne defend pas les différents avis plus anciens! il faudrait ajouter quelques valeurs pour plus de professionalisme! Merci
This is a appropriate blog for everyone who hopes to learn about this topic. You already know a lot its practically challenging to argue with you (not that I just would want…HaHa). You definitely put a whole new spin with a topic thats been revealed for some time. Fantastic stuff, just excellent!
In case you had a parrot, which phrase or phrase would you educate it first?
Great post! We are linking to this particularly great post on our website. Keep up the great writing.
Hello! I simply would wish to supply a huge thumbs up for the wonderful info you might have here for this post. I am returning to your blog post for additional soon.
Naturally I like your web-site, but you have to take a look at the spelling on quite a few of your posts. A number of them are rife with spelling issues and I find it very bothersome to tell you. On the other hand I’ll surely come back again!
It’s my job to dont usually post on many Blogs, still I recently has to say thank you maintain the astonishing work. Ok unfortunately it is time to get at school.
Loving the information on this website, you have done outstanding job on the blog posts.
Having read this I thought it was really informative. I appreciate you spending some time and effort to put this information together. I once again find myself personally spending a significant amount of time both reading and leaving comments. But so what, it was still worth it.
I need to to thank you for this fantastic read!! I definitely enjoyed every little bit of it. I’ve got you book marked to look at new things you post…
Many thanks for creating this place wonderful, i need to say i appreciate all of the exertions that you just do on this great website.
Merely wanna remark on few general things, The website pattern is perfect, the subject matter is really excellent : D.
I discovered your blog web site on google and appearance a couple of your early posts. Always keep in the very good operate. I recently extra up your Feed to my MSN News Reader. Looking for toward reading a lot more by you at a later date!…
Useful information. Fortunate me I discovered your site by accident, and I’m stunned why this coincidence didn’t took place earlier!
I bookmarked it.
you have a wonderful weblog here! do you wish to have the invite posts on my own blog?
I’ve learn several just right stuff here. Certainly value bookmarking for revisiting. I surprise how much effort you put to create the sort of excellent informative web site.
i gave my girlfriend a diamond bracelt and she was quite pleased with it”
This blog was… how do I say it? Relevant!! Finally I’ve found something which helped me. Kudos!
The very next time I read a blog, I hope that it doesn’t fail me just as much as this particular one. I mean, I know it was my choice to read through, nonetheless I actually thought you would have something useful to talk about. All I hear is a bunch of crying about something that you could fix if you weren’t too busy looking for attention.
I seriously love your website.. Excellent colors & theme. Did you create this site yourself? Please reply back as I’m attempting to create my own personal website and would like to learn where you got this from or just what the theme is named. Thank you.
Good information. Lucky me I found your website by accident (stumbleupon). I’ve saved as a favorite for later.
there are bargain dvd players that are sold in our area. i think they are generic low cost dvd players;;
I am glad for writing to make you understand of the beneficial discovery my wife’s princess encountered checking your web site. She realized some details, including how it is like to possess a marvelous giving spirit to have the mediocre ones clearly gain knowledge of a number of tricky subject matter. You truly exceeded people’s desires. Thanks for coming up with the beneficial, trustworthy, informative and as well as easy tips on the topic to Evelyn.
You created some decent points there. I looked online for the issue and located most people is going as well as with your web site.
You really make it appear so easy together with your presentation however I find this topic to
be really one thing which I believe I would never understand.
It seems too complicated and very vast for me.
I’m looking forward in your subsequent post, I’ll attempt to get
the dangle of it!
Decent blog page! That i absolutely love the simplest way it’s easy on my own view as well as information and facts are well written. Now i’m wanting to know earn money may perhaps be warned whenever a new place has been produced. I have subscribed to your own rss feed which inturn have to the secret! Possess a good daytime!
You seem to be very professional in the way you write.*”~*~
Absolutely composed subject material , Really enjoyed reading through .
Awesome read , I am going to spend more time learning about this subject
Aw, this became a very good post. In concept I have to place in writing similar to this moreover – spending time and actual effort to have a good article… but what can I say… I procrastinate alot and also by no means find a way to go done.
Excellent read, I just passed this onto a friend who was doing a little research on that. And he just bought me lunch because I found it for him smile Therefore let me rephrase that: Thank you for lunch!
You live, you battle on, you do it for others, and you help others do so.
What sort of volunteer work do you assume can be essentially the most rewarding for you?
After looking over a handful of the articles on your web page, I seriously appreciate your technique of blogging. I book-marked it to my bookmark website list and will be checking back soon. Please visit my web site as well and tell me what you think.
Excellent article! We will be linking to this particularly great post on our website. Keep up the good writing.
The ultimate game ended on March 29, 1886 when Zukertort tendered his resignation and congratulated the brand new World Champion.
There’s definately a great deal to find out about this subject. I like all the points you made.
This is the proper weblog for anyone who wishes to be familiar with this topic. You know a whole lot its practically challenging to argue on hand (not that When i would want…HaHa). You definitely put a brand new spin on the topic thats been discussing for several years. Great stuff, just wonderful!
There is noticeably a bundle to know about this. I assume you have made certain nice points in functions also.
I can see that you are an somebody in this matter. I am beginning a site soon, and your information will be very effective for me.. Thanks for all your help and wishing you all the prosperity in your business.
Pretty! This has been an incredibly wonderful post. Thank you for providing this info.
For an area-increasing look, you can lengthen these materials to the rest of the bath or add curiosity with completely different wall therapies.
howdy fellow web master! I really like your website! I liked the make of your sidebar.
I like this site very much, Its a rattling nice place to read and get info .
I’m impressed, I must say. Really rarely must i encounter a weblog that’s both educative and entertaining, and without a doubt, you’ve got hit the nail within the head. Your thought is outstanding; the thing is something which inadequate persons are speaking intelligently about. I will be very happy i always stumbled across this during my find something about it.
After looking into a few of the blog articles on your blog, I really appreciate your way of blogging. I saved it to my bookmark site list and will be checking back in the near future. Please check out my website as well and tell me your opinion.
Wonderful post! We will be linking to this particularly great content on our website. Keep up the good writing.
This is a topic which is near to my heart… Thank you! Exactly where are your contact details though?
There are ways to avoid wasting money and nonetheless make sure the trip is comfy on your workers; a travel manager is accessible to plan all areas of your trip successfully and get you wonderful offers.
Thank your for all the enthusiasm to offer such handy information here.
Saved as a favorite, I like your site.
The next time I read a blog, Hopefully it does not disappoint me as much as this one. I mean, Yes, it was my choice to read through, nonetheless I truly believed you would have something interesting to talk about. All I hear is a bunch of moaning about something that you could possibly fix if you were not too busy searching for attention.
Excellent article! We will be linking to this great post on our website. Keep up the great writing.
After exploring a number of the blog articles on your website, I really like your technique of blogging. I added it to my bookmark website list and will be checking back soon. Please visit my web site too and tell me your opinion.
May I simply say what a comfort to find someone who genuinely knows what they are discussing on the net. You certainly understand how to bring an issue to light and make it important. A lot more people need to look at this and understand this side of the story. I was surprised you aren’t more popular given that you most certainly have the gift.
This kind of publish appears to get yourself a lot of visitors. How will you acquire traffic to that? It provides a great unique twist upon issues. I guess having something traditional or perhaps substantial to give info on is the central aspect.
I have been meaning to create about something similar to this on a single of my blogs and this provided a concept. Thanks.
You can increase your blog visitors by having a fan page on facebook.~’:~’
An outstanding share! I’ve just forwarded this onto a friend who was conducting a little research on this. And he in fact bought me lunch due to the fact that I discovered it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanks for spending some time to talk about this matter here on your blog.
Hi! I could have sworn I’ve visited this web site before but after looking at some of the articles I realized it’s new to me. Regardless, I’m certainly pleased I came across it and I’ll be book-marking it and checking back frequently!
The very next time I read a blog, Hopefully it won’t fail me as much as this one. After all, Yes, it was my choice to read, but I truly thought you would have something helpful to say. All I hear is a bunch of complaining about something you could fix if you weren’t too busy seeking attention.
Hi there! This post could not be written any better! Going through this post reminds me of my previous roommate! He constantly kept talking about this. I will send this article to him. Fairly certain he’ll have a good read. Many thanks for sharing!
As I site possessor I believe the content matter here is rattling magnificent , appreciate it for your efforts. You should keep it up forever! Good Luck.
Well done! I appreciate your blog post to this matter. It has been insightful. my blog: how to get rid of love handles
i would love to sip a cup of green tea each morning because it contains L-theanine which calms the mind**
After going over a number of the articles on your site, I seriously like your way of blogging. I bookmarked it to my bookmark website list and will be checking back soon. Please check out my web site as well and tell me what you think.
Howdy! This article couldn’t be written any better! Looking through this article reminds me of my previous roommate! He continually kept preaching about this. I am going to send this post to him. Pretty sure he’ll have a great read. I appreciate you for sharing!
The new smoking alternative has become famouse in the market. It is called the Electronic Cigarettes, bring a packet to your home and than say me how much you liked it.
This is actually serious, You’re a particularly professional blogger. I have signed up with your feed furthermore watch for enjoying all of your incredible write-ups. In addition, I’ve got shared your webpage with our social networks.
Hello! I simply would like to make a enormous thumbs up for your great info you’ve got here for this post. I will be returning to your website to get more detailed soon.
You have brought up a very excellent details , thankyou for the post. “Beginnings are apt to be shadowy and so it is the beginnings of the great mother life, the sea.” by Rachel Carson.
Youre so cool! I dont suppose Ive read anything this way prior to. So nice to find somebody with a few original applying for grants this subject. realy we appreciate you beginning this up. this web site is one area that is required over the internet, somebody if we do originality. useful purpose of bringing something new to the net!
Incredible! This blog looks exactly like my old one! It’s on a entirely different subject but it has pretty much the same page layout and design. Wonderful choice of colors!
I have been reading out some of your stories and i can claim pretty nice stuff. I will surely bookmark your blog.
Aw, this is an incredibly nice post. In thought I must set up writing this way moreover – spending time and actual effort to manufacture a good article… but what / things I say… I procrastinate alot and by no indicates apparently go accomplished.
We’re a group of volunteers and opening a new scheme in our community. Your web site offered us with valuable info to work on. You have done an impressive job and our entire community will be thankful to you.
Pretty! This has been an incredibly wonderful post. Thanks for providing this information.
Your style is very unique in comparison to other people I have read stuff from. Many thanks for posting when you’ve got the opportunity, Guess I will just bookmark this site.
Way cool! Some extremely valid points! I appreciate you writing this article and also the rest of the site is also very good.
You made some decent points there. I looked on the net for the problem and discovered most individuals is going together with together with your internet site.
Nice post. I understand some thing much harder on different blogs everyday. It will always be stimulating to study content from other writers and practice a specific thing from their site. I’d would rather apply certain with all the content on my own weblog whether or not you don’t mind. Natually I’ll offer you a link in your web blog. Many thanks sharing.
I’d have to check with you here. Which isn’t something Which i do! I quite like reading a post that can make people believe. Also, thanks for allowing me to comment!
Excellent write-up. I absolutely appreciate this site. Stick with it!
You should be a part of a contest for one of the most useful sites on the net. I most certainly will recommend this blog!
The Jap Front of the European Theatre of World War II encompassed the battle in central and japanese Europe from 22 June 1941, to 9 Could 1945.
Greetings! Very useful advice in this particular article! It’s the little changes that make the most important changes. Many thanks for sharing!
Pretty! This was an incredibly wonderful post. Thanks for providing this information.
Greetings! Very useful advice in this particular post! It is the little changes that will make the largest changes. Thanks for sharing!
This blog was… how do you say it? Relevant!! Finally I’ve found something which helped me. Thank you.
Hi! I could have sworn I’ve visited this site before but after looking at some of the posts I realized it’s new to me. Anyhow, I’m certainly delighted I came across it and I’ll be bookmarking it and checking back regularly!
This is a very good tip especially to those new to the blogosphere. Simple but very accurate info… Thank you for sharing this one. A must read article.
She thinks it is malfunctioning before she is attacked by it.
You made some good points there. I looked on the net for additional information about the issue and found most people will go along with your views on this site.
Everything is very open with a precise explanation of the challenges. It was really informative. Your site is extremely helpful. Thank you for sharing.
It’s nearly impossible to find experienced people on this subject, but you sound like you know what you’re talking about! Thanks
Oh my goodness! Amazing article dude! Thank you, However I am encountering issues with your RSS. I don’t know the reason why I am unable to subscribe to it. Is there anyone else getting similar RSS problems? Anyone who knows the answer will you kindly respond? Thanx!
Excellent article! We are linking to this particularly great content on our website. Keep up the good writing.
I need to to thank you for this good read!! I definitely loved every little bit of it. I have you saved as a favorite to look at new things you post…
Hello my loved one! I wish to say that this article is amazing, nice written and come with almost all significant infos. I’d like to see extra posts like this .
Very good written article. It will be supportive to anyone who utilizes it, including me. Keep doing what you are doing – can’r wait to read more posts.
Good aftie, i am a blogger too” and i can see that you are a nice blogger too,
A motivating discussion is worth comment. I believe that you need to publish more on this subject, it might not be a taboo matter but usually people don’t talk about these subjects. To the next! Many thanks.
There is certainly a great deal to learn about this subject. I like all of the points you have made.
Spot on with this write-up, I actually believe this amazing site needs a great deal more attention. I’ll probably be back again to see more, thanks for the info!
I like your website.. great shades & motif. Performed people design and style this fabulous website yourself or even have people rely on someone else to make it work for you personally? Plz interact when I!|m looking to pattern by myself blog site plus would want to find out wherever u received this particular through. thank you
you have got a fantastic weblog here! want to make some invite posts on my small blog?
nicotine can really make you an addict, stay away from cigarettes in the first place-
I’m amazed, I have to admit. Rarely do I encounter a blog that’s equally educative and engaging, and without a doubt, you have hit the nail on the head. The problem is something which not enough folks are speaking intelligently about. I’m very happy I stumbled across this in my search for something regarding this.
Next time I read a blog, I hope that it doesn’t fail me as much as this particular one. I mean, Yes, it was my choice to read, but I actually thought you would have something interesting to talk about. All I hear is a bunch of moaning about something you could possibly fix if you were not too busy looking for attention.
I really like it when folks come together and share views. Great site, keep it up!
I blog frequently and I truly appreciate your content. This article has truly peaked my interest. I will book mark your blog and keep checking for new information about once per week. I subscribed to your RSS feed as well.
I really like gathering useful info, this post has got me even more info!
When visiting blogs, i always look for a very nice content like yours ,
Man that was very entertaining and at the same time informative..,*,`
I’d like to thank you for the efforts you have put in penning this website. I’m hoping to see the same high-grade content by you later on as well. In fact, your creative writing abilities has inspired me to get my very own blog now 😉
An fascinating discussion might be priced at comment. I’m sure that you simply write much more about this topic, it might not be a taboo subject but generally individuals are inadequate to dicuss on such topics. To another. Cheers
I must show some appreciation to you for bailing me out of this particular dilemma. As a result of looking throughout the internet and getting techniques that were not beneficial, I thought my entire life was gone. Living without the answers to the difficulties you’ve fixed as a result of your entire post is a serious case, as well as those that would have badly damaged my career if I hadn’t noticed your web blog. Your main mastery and kindness in maneuvering the whole lot was very helpful. I am not sure what I would have done if I hadn’t encountered such a stuff like this. I can at this moment look ahead to my future. Thanks a lot very much for your skilled and result oriented guide. I will not be reluctant to suggest your blog to any person who would like recommendations on this subject.
Just blog walking and want to say hi to the owner, Im enjoying reading your review/story thanks
You created some decent points there. I looked on the internet for any issue and found most people should go as well as with the internet site.
Really clean internet site , appreciate it for this post.
I really love the way you discuss this kind of topic..”\”*.
Good day! Do you know if they make any plugins to help with SEO?
I’m trying to get my website to rank for some targeted keywords
but I’m not seeing very good gains. If you know of any please share.
Thanks! I saw similar article here: Coaching
I do believe all of the concepts you have introduced in your post. They’re very convincing and will definitely work. Nonetheless, the posts are very quick for newbies. May just you please extend them a bit from next time? Thank you for the post.
Regards for just a wonderful knowledge together with in all areas of your webblog, here are some is a touch ask for an internet site subscribers. Who also quotes down the page premium? . . . .Speak out delicately and in addition have a top staff; you’ll turn out some distance.
This is a topic close to my heart cheers, do you have a RSS feed I can use?
Everything is very open with a clear description of the challenges. It was really informative. Your website is very helpful. Many thanks for sharing!
Good day! I could have sworn I’ve been to this site before but after browsing through many of the posts I realized it’s new to me. Anyhow, I’m certainly delighted I found it and I’ll be bookmarking it and checking back frequently.
Greetings! Very useful advice within this post! It’s the little changes that make the greatest changes. Many thanks for sharing!
This excellent website really has all of the info I needed about this subject and didn’t know who to ask.
This is a topic which is close to my heart… Take care! Exactly where can I find the contact details for questions?
Hola! Praising the vibe—it’s brilliant. In fact, the aesthetics brings a outstanding touch to the overall vibe. Looking forward to more!
Hello! Do you know if they make any plugins to help with SEO?
I’m trying to get my site to rank for some targeted keywords but I’m not seeing very good success.
If you know of any please share. Kudos! You can read similar blog here: Code of
destiny
One of the things I enjoy regarding reading websites such as this, is that there aren’t any spelling or lexical errors! Causes it to be tough about the readers sometimes. Very good work upon that and also the subject of this website. Many thanks!
Great web site. Plenty of useful information here. I am sending it to several buddies ans also sharing in delicious. And naturally, thanks for your sweat!
Hi there! I know this is kinda off topic however , I’d figured I’d ask. Would you be interested in trading links or maybe guest authoring a blog article or vice-versa? My site discusses a lot of the same subjects as yours and I believe we could greatly benefit from each other. If you’re interested feel free to shoot me an email. I look forward to hearing from you! Awesome blog by the way!
When I initially left a comment I appear to have clicked on the -Notify me when new comments are added- checkbox and from now on every time a comment is added I get four emails with the exact same comment. Is there an easy method you can remove me from that service? Thanks.
I do believe all the ideas you have presented for your post. They’re very convincing and can definitely work. Still, the posts are very quick for novices. Could you please lengthen them a little from next time? Thank you for the post.
If you don’t wanna see this video click on the next one…. . . It is a person who cares about another human being. about someone who been trough so much and we only ‘love’ when she is thin, soccermom, performer, miss perfect. and when she isn’t men only have hate for her. . it’s so easy to take people down when they already are vulnerable. I call it bullying
Can anyone help me out? It will be much appreciated.
You have observed very interesting details! ps decent internet site.
Simply wanna input that you have a very nice website , I enjoy the pattern it really stands out.
Howdy! I simply wish to offer you a huge thumbs up for your excellent info you have got right here on this post. I am coming back to your web site for more soon.
Bonjour! Grateful for the approach—it’s superb. In fact, the details brings a nice touch to the overall vibe. Keep it up!
Hi there! I could have sworn I’ve been to this website before but after browsing through many of the posts I realized it’s new to me. Nonetheless, I’m certainly pleased I came across it and I’ll be book-marking it and checking back often.
Good Post, I am a big believer in writing comments on websites to inform the blog writers know that they’ve added some thing worthwhile to the world wide web!
I’m curious to find out what blog platform you’re working with? I’m having some small security issues with my latest website and I’d like to find something more safe. Do you have any recommendations?
I just put the link of your blog on my Facebook Wall. very nice blog indeed..”..:
Hi there, i just thought i would publish and now let you know your sites style is really smudged within the K-Melon browser. Anyhow maintain in the very good work.
Yeah, i agree with what was explained in your blog. Thank you for all the info. and your hard work.
You really should be a part of a contest first of the most effective blogs over the internet. I am going to suggest this blog!
In the middle the talking starts and two advantages come to play for the side of the good guys, it will be a cliché sci-fi movie.
Good day! Do you use Twitter? I’d like to follow you if that would be ok. I’m undoubtedly enjoying your blog and look forward to new posts.
Hi there, I found your blog by means of Google at the same time as searching for a related matter, your website got here up, it seems to be great. I have bookmarked it in my google bookmarks.
Right here is the right web site for anybody who wishes to understand this topic. You understand so much its almost tough to argue with you (not that I actually will need to…HaHa). You certainly put a brand new spin on a topic that’s been discussed for years. Great stuff, just wonderful.
You should be a part of a contest for one of the most useful blogs on the net. I most certainly will highly recommend this web site!
I blog frequently and I genuinely appreciate your content. This article has really peaked my interest. I will take a note of your website and keep checking for new details about once per week. I subscribed to your RSS feed too.
Howdy! I just wish to offer you a huge thumbs up for your excellent information you’ve got here on this post. I am coming back to your site for more soon.
As I web-site possessor I believe the content matter here is rattling wonderful , appreciate it for your efforts. You should keep it up forever! Good Luck.
I and my buddies were analyzing the nice information found on your site while at once came up with an awful feeling I never thanked you for those techniques. Those boys were as a result glad to see them and have in effect in actuality been tapping into them. Many thanks for getting simply considerate as well as for opting for this form of impressive issues millions of individuals are really desperate to be aware of. My honest regret for not expressing appreciation to sooner.
You really should experience a tournament first of the finest blogs on the internet. I’m going to suggest this website!
Everything is very open with a very clear explanation of the issues. It was definitely informative. Your website is very useful. Thanks for sharing!
Everything is very open with a precise clarification of the issues. It was really informative. Your website is very helpful. Thanks for sharing.
Hi there! I just wish to give you a huge thumbs up for the excellent information you’ve got here on this post. I am returning to your blog for more soon.
Very good article. I will be dealing with some of these issues as well..
you got to work hard to earn lots of money because it is not very easy to earn,,
I thought it was going to be some boring old publish, but it really compensated for my time. I will publish a link to this page on my blog. I am confident my visitors will discover that quite useful
Im no professional, but I imagine you just crafted an excellent point. You naturally understand what youre speaking about, and I can truly get behind that. Thanks for staying so upfront and so honest.
Very good post. I will be experiencing a few of these issues as well..
I have to thank you for the efforts you’ve put in writing this site. I’m hoping to check out the same high-grade blog posts by you in the future as well. In fact, your creative writing abilities has motivated me to get my own site now 😉
There’s noticeably a bundle to learn about this. I assume you made sure good points in options also.
Zsa zsa Janio speck Patience pupinyo preble vainglory inhaler aguillar
Hi there, Could I grab this post photo and implement that on my own webpage?
It is in reality a great and useful piece
of info. I am satisfied that you simply shared this helpful information with us.
Please keep us up to date like this. Thank you for
sharing.
Learn all about News is very much imptortant to us.
healthier hair is of course mainly due to genetics but food supplementation can also help you get it;
Outstanding post, I think blog owners should larn a lot from this blog its really user genial .
Chile: As of 23 November 2020, Chile is formally open for tourism.
Hi, I do believe this is an excellent blog. I stumbledupon it 😉 I will return yet again since I book-marked it. Money and freedom is the greatest way to change, may you be rich and continue to help other people.
Auto index which witnessed good promoting stress in yesterday’s trade is witnessing some shopping for interest to commerce in inexperienced gaining multiple proportion points.
I’m excited to discover this great site. I need to to thank you for your time due to this fantastic read!! I definitely savored every little bit of it and I have you saved as a favorite to look at new stuff in your website.
It’s hard to come by knowledgeable people in this particular subject, but you seem like you know what you’re talking about! Thanks
Randolph Warburton, Grade 5 Officer, Selby Employment Trade, Ministry of Labour & Nationwide Service.
Howdy! This post couldn’t be written much better! Going through this article reminds me of my previous roommate! He constantly kept talking about this. I am going to forward this information to him. Pretty sure he’ll have a good read. Thank you for sharing!
The explanation I write this now is because I would like others like me, who wrestle with anorexia nervosa or different types of self-abuse, to know that that there’s always hope.
There are actually a number of particulars like that to take into consideration. That could be a great point to deliver up. I offer the ideas above as common inspiration but clearly there are questions just like the one you bring up where the most important factor can be working in sincere good faith. I don?t know if best practices have emerged round issues like that, but I am positive that your job is clearly identified as a fair game. Each boys and girls feel the influence of only a second’s pleasure, for the remainder of their lives.
I do agree with all the ideas you have offered to your post. They are very convincing and can definitely work. Still, the posts are very quick for beginners. Could you please extend them a bit from subsequent time? Thank you for the post.
I couldn’t resist commenting. Well written!
I’ve found your blog before, but I’ve never left a comment. Today, I thought to myself, “I should leave a comment.” So here’s my comment! Continue with the awesome work! I enjoy your articles and would hate to see them end.
Pretty! This was a really wonderful article. Many thanks for supplying these details.
Excellent post. I am going through some of these issues as well..
Henry Clay Alexander was an American banker who served as president, chairman, and CEO of J.P.
Make sure that to break up them in earlier than starting a very faraway trip.
The very next time I read a blog, I hope that it does not disappoint me just as much as this particular one. After all, Yes, it was my choice to read through, however I really believed you’d have something interesting to talk about. All I hear is a bunch of crying about something that you could possibly fix if you weren’t too busy seeking attention.
If the implicit curve consists of a number of elements it must be began several occasions with suitable beginning factors.
Marcia Hopkins Mackie, OBE, Chairman, Northern Eire Hospitals Authority.
The FA have also moved in direction of offering friends with a food buffet instead of the same old sit-down meal forward of kick-off.
Your style is really unique in comparison to other people I’ve read stuff from. Thank you for posting when you’ve got the opportunity, Guess I will just bookmark this page.
I like the layout of your blog and Im going to do the same for mine. Do you have any tips? Please PM ME.
Hi there! I know this is kinda off topic but I was wondering which blog platform are you using for this website? I’m getting sick and tired of WordPress because I’ve had issues with hackers and I’m looking at alternatives for another platform. I would be awesome if you could point me in the direction of a good platform.
Resources similar to the one you mentioned here is going to be really helpful to myself! I will post a hyperlink to this page on my personal blog. I am
Consequently of getting less gear and provides, alpine-style groups want to complete their climbing route in days and it’s thus thought-about a riskier form of mountaineering (e.g.
Bonjour! I love the execution—it’s remarkable. In fact, the execution brings a exceptional touch to the overall vibe. Really inspiring!
After looking into a number of the blog articles on your site, I truly appreciate your technique of writing a blog. I book-marked it to my bookmark webpage list and will be checking back soon. Please visit my website too and tell me your opinion.
Excellent post! We will be linking to this great article on our site. Keep up the good writing.
Neglect that previous wives’ tale about not coaching dogs until they’re six months old.
When you go right into a joint enterprise with another individual, you’ve got investment properties worth 200,000 and so do they, you have doubled your capability.
Bonjour! Delighted by this idea—it’s remarkable. In fact, this post brings a brilliant touch to the overall vibe. Awesome job!
I discovered your site site on the internet and check some of your early posts. Continue to keep up the top notch operate. I recently additional increase your Feed to my MSN News Reader. Seeking forward to reading much more from you finding out later on!…
you have a great blog here! do you need to cook some invite posts on my small blog?
There are some fascinating closing dates on this article but I don’t know if I see all of them middle to heart. There’s some validity however I’ll take maintain opinion until I look into it further. Good article , thanks and we want extra! Added to FeedBurner as effectively
I blog frequently and I genuinely appreciate your content. This article has really peaked my interest. I am going to take a note of your blog and keep checking for new details about once a week. I opted in for your Feed as well.
You should take part in a contest for one of the most useful blogs on the web. I most certainly will recommend this web site!
With our head office in South East London, we are well positioned to handle queries from around London and
the Home Counties.
Awsome site! I am loving it!! Will be back later to read some more. I am taking your feeds also
There are several good techniques for incorporating your danger management methods.
Generate a very boys and girls? Track down stride ritual coupons available for you small children to footwear. Save your dough. Designs, a person contribution ended up being fabulous.
An interesting discussion is definitely worth comment. There’s no doubt that that you ought to write more on this subject, it may not be a taboo matter but usually people don’t discuss such issues. To the next! Kind regards.
The COVID-19 pandemic within the United States has led to the very best variety of whole infections and deaths of any country, though per capita it isn’t the highest.
Right here is the perfect blog for everyone who hopes to find out about this topic. You realize a whole lot its almost tough to argue with you (not that I actually will need to…HaHa). You certainly put a brand new spin on a topic that’s been written about for years. Great stuff, just great.
You may select to view the calendar by day, week, month or a view that presents simply the subsequent four days.
Good site you have here.. It’s hard to find high-quality writing like yours nowadays. I truly appreciate individuals like you! Take care!!
Very good article. I’m facing a few of these issues as well..
Robert Henry Currell, Secretary and Clerk to the Governors, Northern Polytechnic and National College of Rubber Technology, London.
Hey – decent blog, just looking around some kind of blogs, seems a quite good platform you are using. I’m currently using WordPress for a few of my sites but planning to change one of them over to a platform similar to yours as a trial run. Anything in particular you’ll recommend about it?
Aw, this was an extremely good post. Spending some time and actual effort to create a top notch article… but what can I say… I hesitate a whole lot and don’t manage to get nearly anything done.
Performing arts, literary arts, visible arts.
Generally I do not read article on blogs, however I wish to say that this write-up very forced me to check out and do so! Your writing taste has been amazed me. Thanks, very great post.
As the key automobile manufacturers are showing off their finest idea and production ideas, scholar designers have presented some intriguing designs of their very own.
I had a woman on the bar inform me she “cherished my perm” as soon as, nope, this is simply my hair lol,’ one annoyed commenter wrote.
I really like reading an article that can make people think. Also, many thanks for allowing for me to comment.
This appears to be a lucrative supply of investment within the Northeast of Brazil.
An outstanding share! I have just forwarded this onto a coworker who has been conducting a little homework on this. And he actually ordered me lunch due to the fact that I discovered it for him… lol. So let me reword this…. Thanks for the meal!! But yeah, thanks for spending some time to talk about this topic here on your site.
A wardrobe change for the night reception was accompanied by a new hairstyle, with the hair being pinned back into a bun that would have taken simply minutes to create.
I’m impressed, I must say. Rarely do I come across a blog that’s both educative and entertaining, and without a doubt, you have hit the nail on the head. The problem is an issue that too few men and women are speaking intelligently about. Now i’m very happy I found this during my search for something relating to this.
This blog was… how do you say it? Relevant!! Finally I have found something which helped me. Thank you!
Great article. I’m dealing with some of these issues as well..
Oh my goodness! an excellent post dude. Thanks a ton Nevertheless I am experiencing problem with ur rss . Do not know why Cannot enroll in it. Will there be everyone getting identical rss difficulty? Anyone who knows kindly respond. Thnkx
Good post. I’m dealing with some of these issues as well..
I was able to find good advice from your content.
The very next time I read a blog, Hopefully it doesn’t fail me just as much as this one. I mean, I know it was my choice to read through, but I really believed you would probably have something helpful to talk about. All I hear is a bunch of moaning about something you could possibly fix if you were not too busy seeking attention.
This is the perfect blog for anybody who wants to understand this topic. You realize so much its almost tough to argue with you (not that I really would want to…HaHa). You definitely put a brand new spin on a subject that has been written about for years. Great stuff, just great.
This excellent website certainly has all the information I needed concerning this subject and didn’t know who to ask.
It seems solely fair that your partner, children, ferret or other legal heirs might money in your IRA.
This excellent website certainly has all the information and facts I wanted concerning this subject and didn’t know who to ask.
The very next time I read a blog, I hope that it won’t fail me as much as this one. After all, Yes, it was my choice to read, however I truly thought you would probably have something helpful to talk about. All I hear is a bunch of moaning about something that you could fix if you weren’t too busy looking for attention.
This is a great tip particularly to those new to the blogosphere. Simple but very precise info… Many thanks for sharing this one. A must read article.
Way cool! Some very valid points! I appreciate you writing this write-up and the rest of the website is also very good.
Howdy! I could have sworn I’ve been to this site before but after looking at some of the articles I realized it’s new to me. Anyhow, I’m definitely delighted I found it and I’ll be bookmarking it and checking back often!
bookmarked!!, I like your web site.
I blog often and I genuinely thank you for your content. Your article has really peaked my interest. I’m going to take a note of your blog and keep checking for new details about once a week. I opted in for your Feed as well.
Saved as a favorite, I like your web site.
This web site definitely has all the information and facts I wanted concerning this subject and didn’t know who to ask.
You should take part in a contest for one of the best websites on the internet. I am going to recommend this site!
These begin with around ₤ 500, not including
installation fees, according to Which?
You’ve made some decent points there. I looked on the web to find out more about the issue and found most people will go along with your views on this web site.
I blog quite often and I truly thank you for your content. This article has truly peaked my interest. I’m going to bookmark your blog and keep checking for new information about once a week. I subscribed to your Feed too.
Very good post. I’m facing some of these issues as well..
This is a great take on the matter, and I’ve seen a similar opinion shared on https://fabets.dev/. It’s clear that this is a well-discussed topic with valid arguments.
I blog frequently and I really appreciate your information. The article has truly peaked my interest. I am going to take a note of your site and keep checking for new information about once per week. I opted in for your Feed too.
Howdy! This blog post could not be written much better! Looking at this article reminds me of my previous roommate! He always kept talking about this. I will forward this article to him. Pretty sure he’s going to have a very good read. Many thanks for sharing!
bookmarked!!, I really like your blog.
Really enjoyed perusing this article. It’s very well-written and packed with useful information. Thanks for providing this.
Fantastic article. I found the information extremely beneficial. Adored the manner you clarified all the points.
I couldn’t refrain from commenting. Well written.
Top quality! Amazing for what I needed.
I tried it and was worth it.
Good day! I could have sworn I’ve been to this website before but after browsing through some of the posts I realized it’s new to me. Anyhow, I’m definitely pleased I discovered it and I’ll be bookmarking it and checking back frequently.
Genuinely appreciated this post. It provided plenty of useful details. Fantastic job on writing this.
Appreciated the details provided in this article. It’s so articulate and packed with helpful details. Excellent effort!
This article is amazing! Packed with valuable details and highly clear. Thanks for sharing this.
Wonderful article. It’s highly well-written and full of valuable details. Thanks for offering this post.
Fast delivery. Amazing quality.
This is wonderful! Filled with valuable details and highly articulate. Thanks for sharing this.
Hi, I do think this is an excellent blog. I stumbledupon it 😉 I’m going to revisit yet again since I book-marked it. Money and freedom is the best way to change, may you be rich and continue to guide others.
Really liked this article. It provided plenty of valuable details. Fantastic work on composing this.
Wonderful entry. It’s highly articulate and packed with useful insight. Many thanks for offering this post.
Hi, I do believe this is an excellent blog. I stumbledupon it 😉 I will revisit once again since i have bookmarked it. Money and freedom is the greatest way to change, may you be rich and continue to help other people.
Way cool! Some extremely valid points! I appreciate you writing this post and the rest of the website is very good.
After I initially left a comment I seem to have clicked on the -Notify me when new comments are added- checkbox and from now on whenever a comment is added I receive 4 emails with the same comment. Perhaps there is an easy method you are able to remove me from that service? Cheers.
Really enjoyed perusing this article. It’s highly clear and full of valuable insight. Many thanks for sharing this.
I couldn’t resist commenting. Very well written!
An impressive share! I have just forwarded this onto a colleague who had been doing a little homework on this. And he actually bought me lunch simply because I found it for him… lol. So allow me to reword this…. Thank YOU for the meal!! But yeah, thanx for spending time to discuss this matter here on your internet site.
I blog often and I genuinely thank you for your information. The article has truly peaked my interest. I am going to book mark your blog and keep checking for new information about once per week. I subscribed to your Feed as well.
This is a topic that is close to my heart… Thank you! Exactly where can I find the contact details for questions?
I would like to thank you for the efforts you have put in writing this website. I really hope to see the same high-grade content from you later on as well. In truth, your creative writing abilities has inspired me to get my own website now 😉
Hiya! Really enjoying the execution—it’s amazing. In fact, the aesthetics brings a impressive touch to the overall vibe. Way to go!
I needed to thank you for this great read!! I absolutely enjoyed every bit of it. I have got you book marked to look at new stuff you post…
Great info. Lucky me I found your site by chance (stumbleupon). I’ve book marked it for later.
I love reading through an article that will make people think. Also, thank you for allowing me to comment.
Hello there! Embracing the layout—it’s brilliant. In fact, the approach brings a charming touch to the overall vibe. You rock!
Oh my goodness! Amazing article dude! Thank you so much, However I am having troubles with your RSS. I don’t know why I cannot subscribe to it. Is there anybody having the same RSS problems? Anyone that knows the answer can you kindly respond? Thanks.
I used to be able to find good information from your articles.
Nice post. I learn something totally new and challenging on websites I stumbleupon on a daily basis. It’s always useful to read articles from other writers and practice a little something from other websites.
Excellent article! We will be linking to this great post on our site. Keep up the great writing.
After checking out a handful of the blog posts on your web site, I honestly appreciate your technique of blogging. I book marked it to my bookmark site list and will be checking back soon. Take a look at my website as well and let me know what you think.
May I just say what a comfort to find someone that really understands what they’re discussing on the internet. You certainly understand how to bring an issue to light and make it important. More and more people should check this out and understand this side of the story. I was surprised that you aren’t more popular because you definitely possess the gift.
Yo! Admiring your art—it’s fantastic. In fact, the layout brings a fantastic touch to the overall vibe. More please!
Loved the insight in this article. It’s highly well-researched and full of useful information. Excellent job!
Your style is very unique in comparison to other folks I have read stuff from. I appreciate you for posting when you have the opportunity, Guess I will just book mark this web site.
bookmarked!!, I really like your blog!
I totally agree with this comment. It’s a view that I’ve come across on Soc88, and I think it’s one worth paying attention to, given the solid arguments behind it.
Thanks for any other excellent post. Where else may just anybody get that type of information in such
an ideal approach of writing? I’ve a presentation next week, and I am at the
look for such information.
This post is highly enlightening. I really appreciated perusing it. The details is extremely arranged and simple to comprehend.
Everything is very open with a very clear description of the issues. It was truly informative. Your website is very helpful. Thanks for sharing!
I needed this today
# Harvard University: A Legacy of Excellence and Innovation
## A Brief History of Harvard University
Founded in 1636, **Harvard University** is the oldest and one of the most prestigious higher education institutions in the United States.
Located in Cambridge, Massachusetts, Harvard has built a global reputation for academic excellence, groundbreaking research, and influential alumni.
From its humble beginnings as a small college established to educate clergy, it has evolved into a world-leading
university that shapes the future across various disciplines.
## Harvard’s Impact on Education and Research
Harvard is synonymous with **innovation and intellectual leadership**.
The university boasts:
– **12 degree-granting schools**, including the renowned **Harvard Business School**,
**Harvard Law School**, and **Harvard Medical School**.
– **A faculty of world-class scholars**, many of whom are Nobel
laureates, Pulitzer Prize winners, and pioneers in their fields.
– **Cutting-edge research**, with Harvard leading initiatives in artificial intelligence, public health,
climate change, and more.
Harvard’s contribution to research is immense, with billions of dollars allocated to scientific
discoveries and technological advancements each year.
## Notable Alumni: The Leaders of Today and Tomorrow
Harvard has produced some of the **most influential figures** in history, spanning politics, business, entertainment, and science.
Among them are:
– **Barack Obama & John F. Kennedy** – Former U.S.
Presidents
– **Mark Zuckerberg & Bill Gates** – Tech visionaries (though Gates
did not graduate)
– **Natalie Portman & Matt Damon** – Hollywood icons
– **Malala Yousafzai** – Nobel Prize-winning activist
The university continues to cultivate future leaders who shape industries and drive global progress.
## Harvard’s Stunning Campus and Iconic Library
Harvard’s campus is a blend of **historical charm and
modern innovation**. With over **200 buildings**, it features:
– The **Harvard Yard**, home to the iconic **John Harvard Statue** (and the famous “three lies” legend).
– The **Widener Library**, one of the largest university libraries
in the world, housing **over 20 million volumes**.
– State-of-the-art research centers, museums, and performing arts venues.
## Harvard Traditions and Student Life
Harvard offers a **rich student experience**, blending academics
with vibrant traditions, including:
– **Housing system:** Students live in one of 12 residential houses, fostering a strong sense of community.
– **Annual Primal Scream:** A unique tradition where students de-stress by running through Harvard Yard before finals!
– **The Harvard-Yale Game:** A historic football rivalry
that unites alumni and students.
With over **450 student organizations**, Harvard students engage in a diverse range of extracurricular
activities, from entrepreneurship to performing arts.
## Harvard’s Global Influence
Beyond academics, Harvard drives change in **global policy,
economics, and technology**. The university’s research impacts
healthcare, sustainability, and artificial intelligence,
with partnerships across industries worldwide. **Harvard’s endowment**, the largest
of any university, allows it to fund scholarships, research, and public initiatives,
ensuring a legacy of impact for generations.
## Conclusion
Harvard University is more than just a school—it’s a **symbol of excellence, innovation, and leadership**.
Its **centuries-old traditions, groundbreaking discoveries, and transformative education** make it one of the most influential institutions in the world.
Whether through its distinguished alumni, pioneering research, or vibrant student life,
Harvard continues to shape the future in profound ways.
Would you like to join the ranks of Harvard’s legendary scholars?
The journey starts with a dream—and an application!
https://www.harvard.edu/
Hi! I could have sworn I’ve visited this website before but after browsing through a few of the posts I realized it’s new to me. Regardless, I’m definitely delighted I found it and I’ll be book-marking it and checking back frequently!
This is a topic that’s near to my heart… Cheers! Exactly where can I find the contact details for questions?
I like it when people come together and share views. Great blog, stick with it.
Howdy! This blog post couldn’t be written any better! Looking at this post reminds me of my previous roommate! He continually kept preaching about this. I’ll forward this information to him. Pretty sure he’ll have a good read. Many thanks for sharing!
This is gold
https://www.adobe.com/
Oh my goodness! Awesome article dude! Thanks, However I am encountering difficulties with your RSS. I don’t know the reason why I can’t join it. Is there anybody else getting similar RSS issues? Anyone who knows the solution can you kindly respond? Thanx!!
鶴の一声
shittings
灯台下暗し
I seriously love your blog.. Pleasant colors & theme. Did you develop this website yourself? Please reply back as I’m attempting to create my own personal website and want to find out where you got this from or just what the theme is called. Cheers.
Everything is very open with a very clear explanation of the challenges. It was really informative. Your site is very helpful. Many thanks for sharing!
Great post, you have pointed out some fantastic details , I too believe this s a very excellent website.
very nice publish, i certainly love this website, keep on it
I used to be able to find good information from your blog posts.
I blog frequently and I genuinely thank you for your content. Your article has really peaked my interest. I’m going to bookmark your website and keep checking for new details about once per week. I opted in for your RSS feed too.
I really like it when individuals come together and share ideas. Great website, continue the good work.
You have made some really good points there. I checked on the net to find out more about the issue and found most individuals will go along with your views on this site.
I blog frequently and I truly appreciate your content. This article has really peaked my interest. I will book mark your website and keep checking for new details about once per week. I opted in for your Feed too.
Great article. I’m going through many of these issues as well..
Hi there! This blog post couldn’t be written much better! Going through this post reminds me of my previous roommate! He continually kept talking about this. I most certainly will forward this information to him. Pretty sure he will have a great read. Many thanks for sharing!
Hi there! This post couldn’t be written any better! Looking through this post reminds me of my previous roommate! He always kept talking about this. I’ll forward this information to him. Fairly certain he’ll have a good read. Thanks for sharing!
Thanks for the write up! Also, just a heads up, your RSS feeds aren’t working. Could you take a look at that?
Oh my goodness! an amazing article dude. Thank you Nevertheless My business is experiencing trouble with ur rss . Do not know why Struggling to sign up for it. Possibly there is any person getting identical rss problem? Anyone who knows kindly respond. Thnkx
This blog was… how do you say it? Relevant!! Finally I’ve found something which helped me. Kudos!
This excellent website really has all the information I needed concerning this subject and didn’t know who to ask.
Welcome to our premier sports streaming site, your ultimate destination for live and on-demand sports content. Whether you’re into football, basketball, baseball, tennis, or esports, we bring you high-quality streams, real-time scores, and expert commentary—all in one place. Stay connected with your favorite teams and never miss a moment of the action, wherever you are.
스포츠중계
Stream Every Game, Anytime, Anywhere! Catch all the live action from your favorite sports with HD quality and zero lag. Join now and never miss a moment of the game again!
스포츠중계
Welcome to our Cryptocurrency Futures Trading Platform!
Trade a wide range of digital assets with leverage and advanced tools designed for both beginners and professionals. Enjoy secure transactions,
real-time market insights, and 24/7 support to enhance your trading experience.
This blog was… how do you say it? Relevant!! Finally I’ve found something that helped me. Appreciate it!
Pretty! This has been a really wonderful article. Thanks for providing these details.
Hi, I do think this is an excellent website. I stumbledupon it 😉 I will revisit once again since I book-marked it. Money and freedom is the best way to change, may you be rich and continue to help others.
this is a great post!
Hello there! This post couldn’t be written any better! Reading through this
post reminds me of my old room mate! He always kept chatting about this.
I will forward this post to him. Pretty sure he will
have a good read. Thank you for sharing!
Feel free to visit my blog post; discover more here
Thought-provoking read. This really made me think.
Well written. Super relevant to what I’m working on.
Thought-provoking read. This really made me think.
this is a great post!
Aw, this was an incredibly good post. Taking a few minutes and actual effort to generate a top notch article… but what can I say… I hesitate a lot and never manage to get nearly anything done.
Great post! Thanks for breaking it down so clearly.
Truly enjoyed this entry. It gave plenty of useful insights. Fantastic work on composing this.
Arrived perfectly. Great product.
Saved as a favorite, I love your site!
Funcional. Recomendo de olhos fechados.
Me surpreendeu! Produto de alta qualidade.
Magnificent beat ! I would like to apprentice at the same
time as you amend your website, how could i subscribe for a blog site?
The account aided me a appropriate deal.
I had been tiny bit acquainted of this your broadcast provided
vibrant clear concept
Here is my homepage – find more
That is a good tip especially to those fresh to the blogosphere. Short but very accurate info… Thank you for sharing this one. A must read post!
Very nice write-up. I certainly love this site. Stick with it!
Pretty! This has been an incredibly wonderful post. Thank you for supplying this information.
exodermin salbe apotheke Nagelpilz
Salbe Erfahrungen – teilt eure!
Very good post. I definitely love this website. Stick with it!
exodermin medikament Erfahrungen – was sagen echte Nutzer?
Everything is very open with a really clear clarification of the issues. It was really informative. Your website is very helpful. Many thanks for sharing.
This site definitely has all of the information and facts I needed about this subject and didn’t know who to ask.
Казино Драгон Мани – мой выбор для вечернего отдыха.
Слоты грузятся быстро, выплаты без задержек.
http://naomi-bistro.ru/
I like it when people get together and share views. Great blog, keep it up!
I appreciate the SAIF Zone’s focus on community and collaboration for businesses.
indian express saif zone
SAIF Zone’s proximity to Al Hamriyah Port is perfect for my trading company.
companies in saif zone sharjah
sms bomber usa Bomber:
the ultimate tool for USA pranks! Flood numbers with ease.
I want to to thank you for this excellent read!! I certainly enjoyed every bit of it. I have got you book-marked to check out new things you post…
saif zone companies job vacancies
блэк спрут
blacksprut сайт
blacksprut зеркала
как зайти на blacksprut зеркала
блэкспрут зеркало вход
I really needed to read this.
Камеры установлены аккуратно,
всё выглядит эстетично.
обслуживание видеонаблюдения
Профессиональная установка, всё аккуратно и надёжно.
монтаж систем безопасности
Система безопасности работает
идеально, доволен.
видеонаблюдение для бизнеса
Hello! I could have sworn I’ve visited your blog before but after looking at some of the posts I realized it’s new to me. Anyways, I’m certainly happy I discovered it and I’ll be book-marking it and checking back often!
Thank you for the auspicious writeup. It in fact was a amusement account it. Look advanced to more added agreeable from you! By the way, how can we communicate?
closing company in saif zone Zone’s
modern offices are great for productivity.
Starting a best business setup company in dubai in Dubai
is ideal for green energy ventures.
I blog quite often and I truly appreciate your information. This great article has truly peaked my interest. I am going to book mark your site and keep checking for new information about once per week. I subscribed to your Feed too.
flying colour business setup setup in Dubai
benefits from a robust legal framework.
Спасибо за статью, узнал много нового о сепарации!
https://online-learning-initiative.org/wiki/index.php/User:GeorgettaOgilvy
Очень нравится подборка новогодних
фильмов зимой!
палач
This is a topic which is near to my heart… Take care! Exactly where can I find the contact details for questions?
This site truly has all the information I wanted concerning this subject and didn’t know who to ask.
Easypayments упрощает оплату с помощью
карт зарубежных банков. Узнайте подробности!
https://ss13.fun/wiki/index.php?title=Otkryt_Kartu_2G
Easypayments – ваш партнер в оформлении зарубежных карт.
Начните сегодня!
https://sun-clinic.co.il/he/question/otkryt-kartu-17u/
Aw, this was a really good post. Spending some time and actual effort to make a superb article… but what can I say… I hesitate a lot and don’t manage to get nearly anything done.
Learn the details here cbet
An outstanding share! I’ve just forwarded this onto a colleague who had been doing a little research on this. And he in fact ordered me dinner simply because I discovered it for him… lol. So allow me to reword this…. Thank YOU for the meal!! But yeah, thanks for spending some time to discuss this matter here on your web page.
Curious about how much does a casino dealer make?
Discover everything you need to know with our expert-approved guides.
We focus on fair gameplay, responsible gambling, and secure
platforms you can trust. Our recommendations include casinos with top-rated bonuses, excellent support,
and real player reviews to help you get the most out
of every experience. Whether you’re new or experienced, we make sure you play smarter with real chances to win and safe environments.
Curious about how to win at the casino with $20?
Discover everything you need to know with our expert-approved guides.
We focus on fair gameplay, responsible gambling, and secure platforms you can trust.
Our recommendations include casinos with top-rated bonuses,
excellent support, and real player reviews to help you get the most out of every experience.
Whether you’re new or experienced, we make sure you play smarter with real chances to win and safe environments.
Searching for reliable answers to how much do casino dealers make?
We provide detailed, trustworthy information backed by real casino experts.
Our goal is to help you enjoy secure gambling with top-tier platforms that value integrity.
Expect transparent gameplay, verified promotions, and
24/7 support when you follow our recommendations.
Join thousands of players choosing fair, exciting, and bonus-rich
environments for their online casino journey.
Want to better understand do disney cruises have casinos?
You’ve come to the right place. We offer honest advice,
updated facts, and casino options that prioritize safety, fairness, and massive rewards.
Each platform we recommend is licensed, secure, and offers bonuses that actually
benefit the players. From welcome packages to ongoing promos, we help you get more value while enjoying fair play and exciting online gaming adventures.
Explore the topic of don laughlin riverside resort and casino and get insider insights that truly matter.
We’re committed to fairness, accuracy, and player-focused content.
All our featured casinos follow strict compliance standards and come loaded with generous sign-up offers and rewards.
Learn how to enjoy gambling safely while taking advantage of
bonuses, high-return games, and trusted services designed for both beginners and professionals alike.
Curious about how many casinos are in las vegas?
Discover everything you need to know with our expert-approved guides.
We focus on fair gameplay, responsible gambling, and
secure platforms you can trust. Our recommendations include casinos with top-rated bonuses,
excellent support, and real player reviews to help you get the most out of every experience.
Whether you’re new or experienced, we make sure you play smarter with real chances to win and safe environments.
Searching for reliable answers to is there a house hedge on schedule 1 casino?
We provide detailed, trustworthy information backed by real casino experts.
Our goal is to help you enjoy secure gambling with top-tier platforms that value integrity.
Expect transparent gameplay, verified promotions, and 24/7 support when you follow our recommendations.
Join thousands of players choosing fair, exciting, and
bonus-rich environments for their online casino journey.
Want to better understand how to play roulette at casino?
You’ve come to the right place. We offer honest advice, updated facts, and casino options that prioritize safety, fairness,
and massive rewards. Each platform we recommend is
licensed, secure, and offers bonuses that actually benefit the players.
From welcome packages to ongoing promos, we help you get
more value while enjoying fair play and exciting online gaming adventures.
Way cool! Some very valid points! I appreciate you writing this article and the rest of the website is extremely good.
Learn the details here https://dendihs.wrenlaboratories.com/pk/?rolex-game-casino
Go ahead and click here https://reshapely.com/pk/?lucky-777-casino-login
Ваш грузовик требует профессионального обслуживания?
Сервисный центр MinskDiesel предлагает:
Ремонт двигателей, КПП, ходовой части
Компьютерную диагностику.
Замену масла и техобслуживание.
Работу с европейскими и азиатскими грузовиками.
Опытные мастера, Оригинальные запчасти, Гарантия на работы
г. Минск
Подробнее на сайте: https://minskdiesel.by/ https://minskdiesel.by/
After looking into a number of the articles on your website, I honestly appreciate your technique of writing a blog. I added it to my bookmark webpage list and will be checking back in the near future. Take a look at my website as well and let me know how you feel.
http://xt8u.top/vn/?hi88-club-com
https://reshapely.com/vn/?ket-pua-sx30
Купить новую Теслу в США — мечта, но аукционы экономичнее.
homepage
Купил Теслу из США, автопилот — это просто фантастика!
web site
Lifestyle optimization cleaning, freed up our entire weekend. Time-saving champions found. Schedule heroes.
https://flex.skander.pro/en/?rio-casino
Lifestyle optimization cleaning, optimized our busy lifestyle. Productivity partners found. Productivity boost.
Мюзиклы на сайте — как бродвейские постановки.
https://itformula.ca/index.php?title=Ruseriya_29W
Жанры на сайте для всех, даже
редкие, вроде нуара.
https://matiri.mx/index.php/question/ruseriya-70p/
Thank you for the good writeup. It in fact was a amusement account it. Look advanced to more added agreeable from you! By the way, how could we communicate?
https://www.tuforocristiano.com/improvement-your-online-payment-needs/
Военные фильмы на сайте — настоящая
история, без прикрас.
https://wiki.tgt.eu.com/index.php?title=Ruseriya_96t
Reliable team excellence, consistent quality never varies. Consistent excellence achieved. Consistent appreciation.
Thank you for the auspicious writeup. It in fact was a amusement account it. Look advanced to more added agreeable from you! By the way, how could we communicate?
Удобно, что можно учиться дистанционно,
сэкономил время.
https://www.wiki.klausbunny.tv/index.php?title=User:BennieKinne
Exceptional Midtown cleaning, perfect for our Upper East Side apartment. This is Manhattan’s finest service. Manhattan excellence.
Как сэкономить на покупке шин для самосвалов оптом?
http://footyindustry.com/wiki/index.php/Asiancatalog_37q
Green cleaning done right, aligns with our values perfectly. Using you for all green needs. Thanks for caring.
Teerex – шины для карьерной техники, выдерживают любые
испытания!
https://ajuda.cyber8.com.br/index.php/Asiancatalog_43L
Hello! I simply would like to give you a huge thumbs up for your excellent information you’ve got here on this post. I am coming back to your blog for more soon.
Professional maintenance cleaning, lets us focus on what matters. This is the best investment. You’re the best.
Expert-level service with skilled professionals, delivering exceptional results every time. We’re booking months ahead for efficiency. We’re thrilled with the results. http://www.lilymaidesnyc.blogcudinti.com/36775020/lily-maids-house-cleaning-service-of-nyc http://www.lilymaidesnyc.blogdon.net/lily-maids-house-cleaning-service-of-nyc-52633991 http://www.lilymaidesnyc.fireblogz.com/67877430/lily-maids-house-cleaning-service-of-nyc http://www.lilymaidesnyc.blogdigy.com/lily-maids-house-cleaning-service-of-nyc-55837723 http://www.lilymaidesnyc.blog5.net/82766740/lily-maids-house-cleaning-service-of-nyc
Excellent results, made our place spotless in no time. Definitely booking again. You guys rock.
Crisis cleaning champions, turned disaster into perfection. Crisis cleaning champions. Panic averted.
Trustworthy regular service, makes busy NYC life manageable. Our home has never looked better. You’re the best.
Pretty! This has been an extremely wonderful post. Thank you for supplying this info.
Individualized service excellence, individual attention given. Personalization perfection. Tailored appreciation.
We recycle all old metal so Beaufort’s landfills stay lighter and your project leaves a greener footprint on the Pacific Northwest we all love. Proper downspout extensions send runoff well past your flowerbeds so you spend weekends gardening instead of dealing with muddy erosion trenches. Beaufort’s notoriously unpredictable rain makes high-capacity seamless gutters an absolute must for any homeowner who wants long‑term protection and peace of mind.
Тканая латунная сетка в
производстве защитных экранов
Тканая латунная сетка для защиты экранов в различных
отраслях производства
Для оптимизации создания экранов, предназначенных для
защиты от различных внешних воздействий, рекомендуется использовать металлические сетки с высокой прочностью и стойкостью к коррозии.
Данный материал сочетает в себе долговечность и функциональность,
что делает его идеальным кандидатом для обеспечения надежной
защиты.
Металлические структуры из бронзы обеспечивают отличный уровень фильтрации, одновременно позволяя воздухопроницаемость, что играет
важную роль в предотвращении накопления тепла.
Такая особенность особенно важна в условиях производственных процессов, где перегрев может привести
не только к ухудшению работы механизмов,
но и к необходимости частого обслуживания оборудования.
При выборе подобного материала стоит обратить внимание
на размер ячеек и толщину проволоки, так как это напрямую влияет на защитные характеристики.
Оптимальный выбор зависит от специфики производственного процесса и требуемого уровня защиты, поэтому рекомендуется провести предварительные испытания на совместимость с другими используемыми элементами системы.
Преимущества латунной сетки для защиты от механических
повреждений
Выбор материалов для защиты оборудования и рабочей среды должен основываться на их прочности и
устойчивости. Использование металлической структуры из меди и цинка
обеспечивает отличные механические характеристики.
Такие изделия выделяются высокой прочностью
на изгиб и удар. Это делает их надёжным барьером против внешних воздействий.
Долговечность материалов также является важной характеристикой.
Обработка с использованием соответствующих антикоррозийных составов значительно увеличивает срок службы.
Это снижает частоту замены, что экономит средства на
пусконаладочные работы.
Механические свойства играют
ключевую роль в предотвращении
повреждений. Сетка демонстрирует хорошую степень деформации, что позволяет ей
поглощать удары без разрушения.
Это делает возможным использование в разнообразных условиях, таких как производственные или промышленные помещения.
Помимо этого, структура обеспечивает отличный уровень защиты от
мелких предметов и частиц.
Это важно для сохранения целостности
оборудования и безопасности персонала,
что особенно актуально в условиях
с высоким уровнем опасности.
С учетом перечисленных фактов, применение
этого материала в решениях
для защиты от внешних угроз становится наиболее оправданным и целесообразным.
Инвестирование в данный сегмент может значительно повысить безопасность и эффективность работы на многих производственных объектах.
Технологические особенности изготовления защитных экранов с использованием тканой латунной сетки
При создании монолитных конструкций важно учитывать выбор ячейнистого материала, способствующего защите от электромагнитного излучения.
Рекомендуется использовать полотно с
высокой плотностью переплетения, что обеспечивает необходимую прочность
и долговечность изделий.
Технология подготовки включает предварительный анализ размеров ячеек, который зависит от предполагаемого спектра воздействия.
Для защиты от высокочастотных волн желательно выбирать варианты с меньшими ячейками,
увеличивая экранирующую способность.
В процессе формовки обратите внимание на точность раскроя и соблюдение геометрии конструкций.
Применение автоматизированных систем позволяет достичь необходимого уровня повторяемости и минимизировать отходы материала.
Слияние элементов может выполняться с
использованием высокотемпературной пайки или сварки, что гарантирует надежность соединений.
Используйте защитные средства, чтобы избежать
окисления соединительных зон, что существенно влияет на долговечность.
После окончательной сборки стоит
провести тестирование на устойчивость к различным типам воздействия.
Применение специализированных методик позволит оценить эффективность, снизить риски поломок и упростить обслуживание
готового изделия.
Для улучшения функциональных характеристик рекомендуется
покрытие антикоррозийными составами.
Это увеличивает срок службы и способствует сохранению
визуально привлекательного вида озвученных защитных конструкций.
Also visit my blog :: https://rms-ekb.ru/catalog/latun/
This is the perfect website for anybody who really wants to understand this topic. You understand so much its almost hard to argue with you (not that I really would want to…HaHa). You certainly put a brand new spin on a topic which has been written about for a long time. Excellent stuff, just excellent.
Emergency response excellence, rescued our reputation completely. Emergency cleaning perfected. Emergency excellence.
Hi there! I could have sworn I’ve visited this website before but after browsing through many of the articles I realized it’s new to me. Anyways, I’m certainly delighted I found it and I’ll be bookmarking it and checking back often!
Every installation comes with stainless steel micro‑mesh guards that laugh at fir needles, keeping maintenance low even during those blustery November storms. With rain coming sideways across Commencement Bay, hidden drip‑edge flashing stops water from sneaking behind the fascia and into your attic insulation. Annual tune‑ups are available; we flush, reseal corners and adjust hangers so your system keeps working even after the roughest winter freeze–thaw cycles.
Primera is a modern supplement designed to support pelvic health. It uses selected natural ingredients to improve bladder control and help urinary tract health while focusing on overall pelvic wellness.
Perfectly customized service, personalization makes the difference. Custom solution providers. Personalized perfection.
This website really has all of the information and facts I wanted about this subject and didn’t know who to ask.
https://storage.googleapis.com/seamlessgutter4less/tacoma-wa/pro-gutter-tips-for-tacomas-wet-climate.html
Thank you, I have recently been searching for info about this topic for a while and yours is the best I’ve discovered till now. However, what in regards to the bottom line? Are you certain about the supply?
Welcome to NanoDefense Pro is the official website of a powerful supplement that is an advanced skincare and nail support formula that uses cutting-edge nanotechnology to rejuvenate and restore health from within.
I must thank you for the efforts you have put in writing this website. I’m hoping to view the same high-grade content by you later on as well. In fact, your creative writing abilities has inspired me to get my own, personal site now 😉
Hey can I get you designers contact info? This is an awesome skin.
I must examine with you here. Which isn’t one thing I often do! I get pleasure from reading a post that may make individuals think. Also, thanks for permitting me to remark!
Hey there just wanted to give you a quick heads up. The words in your article seem to be running off the screen in Safari. I’m not sure if this is a format issue or something to do with web browser compatibility but I figured I’d post to let you know. The style and design look great though! Hope you get the problem solved soon. Thanks
when taking your watch for a repair, always look for a reputable and experienced watch repairman,
Good ¡V I should certainly pronounce, impressed with your site. I had no trouble navigating through all the tabs and related info ended up being truly easy to do to access. I recently found what I hoped for before you know it in the least. Quite unusual. Is likely to appreciate it for those who add forums or something, site theme . a tones way for your customer to communicate. Nice task..
I simply had to thank you so much again. I do not know the things that I would have tried without the type of pointers discussed by you over this theme. It previously was a very scary crisis in my view, however , noticing the very skilled technique you managed that forced me to jump over contentment. I am just happier for this support and thus expect you know what a great job you’re providing instructing people by way of your blog post. Probably you haven’t got to know any of us.
*Nice post. I learn something more challenging on different blogs everyday. It will always be stimulating to read content from other writers and practice a little something from their store. I’d prefer to use some with the content on my blog whether you don’t mind. Natually I’ll give you a link on your web blog. Thanks for sharing.
I impressed, I must say. Really rarely do I encounter a blog that both educative and entertaining, and let me inform you, you’ve got hit the nail on the head. Your thought is outstanding; the issue is something that not sufficient people are speaking intelligently about. I’m very pleased that I stumbled throughout this in my seek for one thing regarding this.
Its like you learn my mind! You appear to grasp a lot
about this, such as you wrote the e book in it or something.
I feel that you can do with some p.c. to drive the message house a bit, however
instead of that, this is wonderful blog. A great read.
I’ll certainly be back.
I don’t even understand how I finished up right here, but I thought this publish was good. I don’t know who you are but certainly you’re going to a well-known blogger if you are not already Cheers!
powerful supplement that is an advanced skincare and nail support formula that uses cutting-edge nanotechnology to rejuvenate and restore health from within.
you are in reality a excellent webmaster. The site
loading speed is incredible. It kind of feels that you are doing any unique trick.
Furthermore, The contents are masterpiece. you have performed a magnificent task in this topic!
hello, i came in to learn about this topic, thanks alot. will put this site into my bookmarks.
Ich kenne einige Leute, die aus Kanadakommen. Eines Tages werde ich auch dorthin reisen Lg Daniela
Image ads are usually only for Adsense regarding Articles internet pages without designed for Adsense intended for Serp’s internet pages.
Launch memecoins Solana without coding skills
https://wiki.lerepair.org/index.php/Utilisateur:KiaraDemarest
https://s3.amazonaws.com/gutterinstallation/tacoma-wa/why-gutters-matter-for-tacoma-homes-in-2025.html
Pumpfun bot makes meme trading simple
http://wiki.envirobatbdm.eu/Utilisateur:RonBaylee970
With rain coming sideways across Commencement Bay, hidden drip‑edge flashing stops water from sneaking behind the fascia and into your attic insulation. Local building codes here in Pierce County require you to manage runoff responsibly, so a properly sloped gutter system keeps foundations safe and neighbors happy. Roll‑forming gutters right in your driveway means each section is one continuous piece—no leaky joints, no wasted metal, no compromises on quality.
nail support formula that uses cutting-edge nanotechnology to rejuvenate and restore health from within.
This web site truly has all of the info I wanted concerning this subject and didn’t know who to ask.
I think this is among the most vital information for me. And i’m glad reading your article. But should remark on few general things, The web site style is ideal, the articles is really nice : D. Good job, cheers
eye doctors are specially helpful whenever you have some eye problems**
Very nice article. I certainly appreciate this site. Keep writing!
I like it whenever people come together and share thoughts. Great blog, keep it up!
After checking out a number of the articles on your site, I honestly appreciate your technique of blogging. I book marked it to my bookmark site list and will be checking back in the near future. Please visit my website as well and tell me how you feel.
We’re a group of volunteers and opening a new scheme in our community.
Your website provided us with valuable information to work on. You have done
an impressive job and our entire community will be grateful to you.
Choosing 6‑inch K‑style aluminum troughs means up to 40 percent more water moves away from your roof, preventing the dreaded waterfall effect in heavy downpours. Local building codes here in Pierce County require you to manage runoff responsibly, so a properly sloped gutter system keeps foundations safe and neighbors happy. Roll‑forming gutters right in your driveway means each section is one continuous piece—no leaky joints, no wasted metal, no compromises on quality.
This is the right website for anyone who hopes to find out about this topic. You know a whole lot its almost hard to argue with you (not that I actually will need to…HaHa). You certainly put a fresh spin on a subject which has been written about for many years. Excellent stuff, just wonderful.
An outstanding share! I’ve just forwarded this onto a co-worker who was conducting a little homework on this. And he actually ordered me dinner simply because I found it for him… lol. So allow me to reword this…. Thanks for the meal!! But yeah, thanks for spending some time to discuss this subject here on your blog.
magnificent issues altogether, you just won a new reader. What may you recommend in regards to your publish that you made some days ago? Any positive?
A fascinating discussion is worth comment. I do think that you should write more about this subject, it may not be a taboo subject but generally people don’t speak about these subjects. To the next! Cheers!
If some one desires to be updated with hottest technologies therefore he must be pay a quick visit this web page and be up to date
all the time.
An outstanding share! I’ve just forwarded this onto a friend who has been doing a little homework on this. And he actually ordered me lunch due to the fact that I discovered it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanx for spending time to talk about this issue here on your website.
My spouse and I absolutely love your blog and find many of your post’s to be exactly I’m looking for. Would you offer guest writers to write content for you personally? I wouldn’t mind composing a post or elaborating on some of the subjects you write regarding here. Again, awesome web log!
I’m not sure where you are getting your information, but good topic. I needs to spend some time learning much more or understanding more. Thanks for great information I was looking for this information for my mission.
The next time I read a blog, Hopefully it doesn’t disappoint me as much as this one. I mean, I know it was my choice to read through, nonetheless I genuinely thought you would have something useful to say. All I hear is a bunch of whining about something you can fix if you weren’t too busy seeking attention.
This web site truly has all of the information and facts I needed concerning this subject and didn’t know who to ask.
I’m very pleased to uncover this web site. I wanted to thank you for ones time for this particularly wonderful read!! I definitely loved every part of it and I have you book-marked to check out new stuff in your website.
Мебель для ванной сделали по нашим размерам,
всё идеально.
интерика мебель сызрань
Pretty! This has been an incredibly wonderful article. Thanks for providing this info.
Прихожая получилась компактной, но
вместительной, супер!
мебель московская сызрань
thanks
Having read this I believed it was really enlightening. I appreciate you taking the time and effort to put this information together. I once again find myself personally spending a significant amount of time both reading and posting comments. But so what, it was still worth it.
Wonderful post! We will be linking to this great content on our website. Keep up the good writing.
You’ve made some really good points there. I checked on the internet for more info about the issue and found most individuals will go along with your views on this web site.
Север Казино покерные турниры и их особенности
Север Казино предлагает захватывающие турниры по покеру для игроков разных уровней
Имея в запасе множество ярких моментов, участие в карточных мероприятиях обладает уникальными преимуществами.
Для начинающих игроков важно обратить внимание на
высокие шансы на успех и обширный выбор различных
форматов, которые предлагают площадки
для аналогичных игр.
Стратегическая глубина и эмоции ожидания – вот что привлекает внимание к
подобным событиям. Поначалу особенно полезно поучаствовать в недорогих или даже бесплатных состязаниях, чтобы понять механизмы
и особенности взаимодействия с другими участниками.
Так можно развить мастерство без значительных финансовых рисков и выиграть полезный
опыт.
Обращая внимание на бонусные программы и специальные акции, важно быть в курсе выгодных предложений учреждения, которые могут значительно увеличить банкролл.
Некоторые площадки применяют систему кэшбэков
или начисляют дополнительные очки за участие в
конкурсах, что делает игру более заманчивой.
На таких мероприятиях обязательно доступны
перекрестные акции и сопроводительные условия, которые
стоит изучить заранее.
Качество игры и разнообразие форматов также являются
немаловажными факторами.
Часто игроки имеют возможность выбирать
между классическими и современными вариантами с
интересными механиками. Поэтому, не упустите шанс ознакомиться с теми предложениями, которые
могут максимально удовлетворить ваши предпочтения и стратегические задумки.
Форматы соревнований на платформе:
чем они отличаются?
В рамках данной платформы предлагаются
различные схемы проведения состязаний, каждая из
которых имеет свои уникальные характеристики и правила.
Основные форматы включают: классические, с ускоренным временем
и слайд-формат.
Классические схемы проходят в привычном темпе,
позволяя игрокам тщательно продумывать свои ходы и стратегии.
Здесь используется стандартный стек фишек, что
обеспечивает плавное развитие событий.
Максимальное время на раздачу даёт возможность участникам анализировать игру соперников.
Формат с ускорением предполагает
более быстрое время на принятие решений.
В этом случае время на ход уменьшается,
что добавляет элемент напряжения.
Игроки должны быть готовы к быстрым действиям, и это требует особой концентрации и навыков.
Обычно такой формат подходит для опытных участников, предпочитающих динамику и активное взаимодействие.
Слайд-формат представляет собой инновационный подход,
где игроки могут зайти, когда захотят, при
этом время и стек изменяются
в зависимости от общих успехов.
Это позволяет новичкам адаптироваться к игре и не чувствовать
давления от фиксированной структуры.
Участники могут в любой
момент присоединиться, что делает
его привлекательным для широкого круга
игроков.
Таким образом, в зависимости от личных предпочтений и уровня опыта, каждый игрок сможет выбрать подходящий для себя формат и насладиться игрой на платформе.
Адаптация стратегий под каждую схему позволяет
максимально эффективно использовать имеющиеся навыки и минимизировать риски.
Стратегии для успешной игры на турнирах
Сосредоточьтесь на bankroll management.
Определите сумму, которую готовы потратить, и придерживайтесь её.
Это поможет избежать эмоциональных решений в процессе игры.
Играйте более агрессивно на ранних стадиях.
В начале состязания ваша
цель – накопить фишки. Используйте возможность делать рейзы и ребайсы для наращивания стека.
Следите за позициями за столом.
Понимание роли позиции в игре – ключ к успеху.
Играйте более рискованно в поздней позиции и будьте осторожнее в ранней.
Часто меняйте стиль игры.
Применяйте тайтовый стиль
против агрессивных оппонентов, и
наоборот. Это поможет сбить соперников с толку.
Изучайте противников. Обратите внимание на их стиль
игры и используйте эту информацию для
принятия решений. Отличное умение адаптироваться к соперникам – залог успеха.
Используйте блеф с умом.
Блеф должен быть целенаправленным.
Выбирайте моменты, когда у вас есть шанс
заставить противника сбросить карты.
Не забывайте про читы в игре.
Сохраняйте спокойствие в напряженных ситуациях и следите за своей мимикой
и жестами, чтобы не выдать свои карты.
Эмоции могут быть вашими противниками.
Планируйте свой выход. Всегда имейте стратегию
на случай, если окажетесь в проигрышной ситуации.
Понимание, когда стоит покинуть игру, может спасти ваш банкролл.
регистрация казино Север
I’m amazed, I have to admit. Seldom do I come across a blog that’s both equally educative and amusing, and let me tell you, you’ve hit the nail on the head. The problem is something that too few men and women are speaking intelligently about. Now i’m very happy that I came across this in my search for something regarding this.
Spot on with this write-up, I truly think this web site wants much more consideration. I’ll most likely be again to learn far more, thanks for that info.
I have to thank you for the efforts you’ve put in writing this site. I really hope to see the same high-grade blog posts by you in the future as well. In truth, your creative writing abilities has motivated me to get my own website now 😉
Бонус daily.
игровой автомат Book of Ra Deluxe 10
thanks
I really love your website.. Great colors & theme. Did you build this web site yourself? Please reply back as I’m attempting to create my own personal site and would love to learn where you got this from or just what the theme is named. Appreciate it!
Минус: лимиты на вывод для новичков, но после верификации все ок.
игровой автомат Gonzo’s Quest Megaways
Депозит бонус 100% — старт мощный.
лучшие бонусы казино
Fantastic post however I was wondering if you could write a litte more on this subject? I’d be very thankful if you could elaborate a little bit further. Many thanks!
May I just say what a comfort to uncover someone that really knows what they are talking about on the net. You definitely know how to bring an issue to light and make it important. More and more people really need to look at this and understand this side of your story. I was surprised that you are not more popular given that you certainly have the gift.
I enjoy looking through an article that can make men and women think. Also, thanks for allowing me to comment.
Next time I read a blog, Hopefully it does not disappoint me just as much as this particular one. After all, Yes, it was my choice to read through, however I actually thought you would have something useful to talk about. All I hear is a bunch of whining about something you could fix if you weren’t too busy seeking attention.
Hi, I believe your blog may be having web browser compatibility issues. When I look at your blog in Safari, it looks fine however when opening in Internet Explorer, it has some overlapping issues. I simply wanted to give you a quick heads up! Other than that, wonderful site.
I really like it when people come together and share views. Great blog, stick with it.
Great post. I am facing many of these issues as well..
Your style is very unique in comparison to other people I have read stuff from.
I appreciate you for posting when you’ve got the opportunity, Guess I’ll just bookmark this web site.
пенсия фсб расчёт
Thanks a lot for sharing this with all of us you actually
recognize what you are speaking about! Bookmarked.
Kindly also consult with my web site =).
We will have a link change contract among
us
Подписчики в ТГ любят бесплатные раздачи
подписчики youtube
Недвижимость в Эмиратах — это не только роскошь,
но и выгодное купить недвижимость в оаэложение средств.
https://s3.amazonaws.com/gutterinstallation/tacoma-wa/tacoma-gutter-innovations-whats-new-in-2025.html
Good post. I learn something new and challenging on blogs I stumbleupon everyday. It’s always exciting to read through articles from other writers and use something from other sites.
very nice publish, i definitely love this web site, keep on it
That is a really good tip particularly to those new to the blogosphere. Simple but very precise info… Thank you for sharing this one. A must read post!
I’m impressed, I must say. Actually hardly ever do I encounter a blog that’s both educative and entertaining, and let me tell you, you’ve got hit the nail on the head. Your thought is excellent; the issue is one thing that not sufficient persons are talking intelligently about. I am very comfortable that I stumbled across this in my seek for one thing regarding this.
Spot on with this write-up, I really believe that this web site needs much more attention. I’ll probably be returning to read through more, thanks for the advice.
I’m really enjoying the design and layout of your website. It’s a very easy on the eyes which makes it much more enjoyable for me to come here
and visit more often. Did you hire out a designer to create your theme?
Great work!
Also visit my homepage … you can try here
I am regular visitor, how are you everybody? This post
posted at this web site is actually pleasant.
my web-site … navigate to this website
I blog often and I really thank you for your information. This great article has truly peaked my interest. I will bookmark your site and keep checking for new information about once per week. I opted in for your RSS feed too.
https://s3.amazonaws.com/gutterinstallation/tacoma-wa/tacoma-gutter-innovations-whats-new-in-2025.html
Everyone loves it when individuals get together and share views.
Great site, continue the good work!
Also visit my webpage hop over to here
Great info. Lucky me I ran across your blog by accident (stumbleupon). I’ve saved as a favorite for later!
I could not refrain from commenting. Exceptionally well written.
This is a good tip particularly to those fresh to the blogosphere. Short but very accurate info… Appreciate your sharing this one. A must read post.
I blog quite often and I truly thank you for your content. This great article has really peaked my interest. I am going to book mark your site and keep checking for new information about once a week. I opted in for your Feed as well.
Военный портал с калькуляторами — всё в одном:
пенсия, ипотека, довольствие.
надбавка за выслугу лет
I blog often and I genuinely appreciate your content. This great article has really peaked my interest. I am going to book mark your site and keep checking for new details about once a week. I subscribed to your Feed as well.
Roll‑forming gutters right in your driveway means each section is one continuous piece—no leaky joints, no wasted metal, no compromises on quality. Because we’re veteran‑owned we show up on time, measure twice, and treat every property with the kind of respect we learned back in basic training. Investing in quality gutters is like buying the good umbrella—it costs a bit more up front but saves you from soaking through your socks every single storm.
I must thank you for the efforts you’ve put in writing this site. I am hoping to see the same high-grade blog posts from you later on as well. In fact, your creative writing abilities has encouraged me to get my own, personal blog now 😉
Great site you have here.. It’s difficult to find high-quality writing like yours these days. I honestly appreciate individuals like you! Take care!!
Milanobet güvenilir mi sorusunun yanıtını bu yazıda size aktardığımız bilgiler kapsamında kendiniz rahatlıkla verebileceksiniz. Öncelikle genel olarak herhangi bir bahis sitesinin güvenilir olup olmadığı hususunda birkaç önemli kriter kullanılmaktadır.
Ahaa, its nice conversation on the topic of this paragraph here at this webpage,
I have read all that, so at this time me also commenting here.
my web page: wikipedia reference
Executive search семинар был насыщенным и полезным.
Рекомендую всем в HR!
курсы хедхантинга
You should be a part of a contest for one of the highest quality sites online. I’m going to highly recommend this site!
Milanobet ile iligli tüm detayları sitemizde bulabilirsiniz.
Thanks for sharing your thoughts about you can find out more.
Regards
Feel free to surf to my blog this site
This website was… how do I say it? Relevant!! Finally I’ve found something which helped me. Thank you!
The Mtaur coin has a dedicated and active wallet. Managing
my assets has never been easier.
My page :: https://t.me/s/minotaurus_official
You need to take part in a contest for one of the best sites on the net. I am going to recommend this site!
You ought to be a part of a contest for one of the greatest websites on the net. I most certainly will recommend this web site!
Ich bin so froh, exodermin wo kaufen entdeckt zu haben! Meine
Hautprobleme sind weg.
exodermin nagelpilz test hat meinen Nagelpilz schnell behandelt.
Ich kann es nur weiterempfehlen!
Estoy encantado con exodermin mercadona.
Mis uñas lucen saludables de nuevo y es fácil de
aplicar.
The next time I read a blog, I hope that it doesn’t fail me just as much as this particular one. I mean, I know it was my choice to read, but I actually thought you would probably have something helpful to say. All I hear is a bunch of moaning about something that you could possibly fix if you weren’t too busy searching for attention.
exodermin dm ha trattato la mia micosi alle unghie in sole due settimane.
Sono entusiasta!
Exodermin m’a beaucoup aidé avec ma mycose des ongles.
Les résultats sont super !
l’exodermin
The Minotaurus mtaur token allocation for
rewards is generous. It really incentivizes active participation.
exodermin ára hat meinen Juckreiz schnell gestoppt.
Die Anwendung ist einfach, und die Ergebnisse
sind top!
Ich benutze exodermin rossmann preis seit einer Woche,
und die Ergebnisse sind fantastisch. Kein Juckreiz mehr!
Hey! I could have sworn I’ve been to this website before but after checking through some of the post I realized it’s new to me.
Nonetheless, I’m definitely happy I found it and I’ll be book-marking and checking back often!
Feel free to surf to my web site … imp source
https://pangudownloads.com/beauty/erectil/
exodermin precio farmacia similares resolvió mis problemas
de piel rápidamente. ¡Estoy muy satisfecho!
Bahis dünyasının en güvenilir sitesine hoşgeldiniz. hemen giriş yapın deneme bonusunuzu alın!
Środek na grzybicę jak Exodermin jest unikalny. Paznokcie wróciły Krem do pielęgnacji stóp normy.
Cena w promocji
Business growth becomes smoother with better Briansclub credit.
https://mercedes-world.com/c-class/mercedes-amg-c-53-spy
exodermin erfahrung hat meine Erwartungen übertroffen. Meine Nägel
sehen wieder gut aus!
La crème exodermin france a résolu mes problèmes de peau.
En une semaine, tout va mieux !
Ich habe exodermin bei nagelpilz in der Apotheke gekauft und bin begeistert!
Der Pilz ist verschwunden.
La aplicación de donde puedo comprar exodermin es muy fácil.
¡Mi piel se siente saludable de nuevo!
Sono entusiasta di exodermin funziona!
La mia pelle è di nuovo sana e il prurito è sparito.
Я однажды сэкономил, но в итоге получил спамные ссылки, которые только
навредили сайту.
Заказать прогон хрумером недорого Хрумером с гарантией — это лучший вариант для тех, кто
ценит результат.
Hol lehet Exodermin olcsón venni Szegeden gyors kiszállítással.
A gombák eltűntek. Az ár kedvezményes
Hondrolife hat meine Morgensteifigkeit reduziert.
Ich starte besser in den Tag.
Gelenkschmerzen Symptome
Противогъбичен крем Exodermin с прополис действа антибактериално.
Използвах го 5 мита за гъбичките по ноктите сърбеж и сега съм свободна
от инфекция. Цената е под 60 лв.
Türkiye’nin en güvenilir bahis sitesi olan Vegabet’e üye olarak deneme bonusu alabilirsin. https://vegabetbahis.com/ sitesi üzerinden üye ol deneme bonusun anında hesabına yüklensin.
This is a very good tip particularly to those new to the blogosphere. Short but very accurate information… Thank you for sharing this one. A must read article!
Pretty! This was an extremely wonderful article. Thanks for providing this information.
This web site really has all the information and facts I wanted concerning this subject and didn’t know who to ask.
This is a topic that is near to my heart… Thank you! Exactly where can I find the contact details for questions?
Greetings! Very useful advice within this post! It is the little changes that make the greatest changes. Many thanks for sharing!
Exodermin w Krakowie w Wrocławiu –
kupiłem tanio. Paznokcie zdrowe. Zamów z oficjalnej
strony
Hondrolife в София е
спасение за хора с хронични болки
Exodermin отстъпка 50% – супер
оферта. Кожата ми е здрава.
Мнения от форуми са топ
Противогъбичен крем за спортисти
An interesting discussion is worth comment. I do believe that you should write more about this issue, it may not be a taboo subject but generally people don’t talk about such topics. To the next! All the best.
Hogyan használd helyesen az Exodermint?
Naponta kétszer tiszta bőrre. A gombák eltűntek
Gombaellenes krém
Öncelikle, kaçak bahis sitelerine üye olmak ve bu sitelerde bahis oynamak, Türk Ceza Kanunu ve ilgili mevzuat kapsamında suç sayılabilir. Bu durum, sadece sitenin sahipleri için değil, aynı zamanda kullanıcılar için de geçerlidir. Yani, bu tür platformlarda oynamak para cezası veya hapis cezası gibi yaptırımlarla sonuçlanabilir. Üstelik, bu tür sitelerde yaşanan dolandırıcılık olayları da mahkemeye taşınabilir ve mağdur olan kullanıcılar haklarını aramakta zorluk çekebilir.
Игровые автоматы бесплатно в Лев
казино — отличная тренировка перед реальной игрой!
https://www.pro-parikmahera.ru/kurs/pages/?sands_sohranili_v_indekse_dow_jones_sustainability_asia_pacific.html
Лучшие онлайн казино России — Лев казино
всегда в лидерах!
http://materinstvo2.com/demo-igry-avtomaty-onlajn-sekrety-besplatnoj-igry-i-pochemu-eto-vazhno-znat-kazhdomu/
Купили LED-экран для презентаций,
и теперь даже самые сложные проекты воспринимаются клиентами с энтузиазмом
https://yogaasanas.science/wiki/User:HiltonWlj6953727
Good blog post. I absolutely appreciate this website. Thanks!
I blog quite often and I really thank you for your information. Your article has really peaked my interest. I am going to take a note of your site and keep checking for new information about once per week. I opted in for your RSS feed as well.
A Hondrolife-t egy barátom ajánlotta, aki szintén használja
Spray artrózisra irodai dolgozóknak
fortunica online’s banking options are super convenient.
Crypto deposits are a breeze.
The VIP program at fortunica casino online is worth it if you’re a regular player.
The rewards are amazing.
Bonus veren siteler arasında seçim yaparken dikkatli olmalı ve hangi sitenin size en fazla faydayı sağlayacağını analiz etmelisiniz. Bazı siteler daha düşük çevrim şartlarına sahip olabilirken, diğerleri daha yüksek bonus miktarları sunabilir. Bu nedenle, kendi oyun ve bahis alışkanlıklarınıza en uygun siteyi seçmek önemlidir.
The VIP program at fortunica casino is definitely worth it if you play regularly.
The rewards are fantastic.
Vegabet’in güncel promosyonları için telegram adresini takip etmeyi unutmayın. https://t.me/VegaResmi
The Big Bass Splash slot at fortunica casino
is so entertaining. The graphics are top-notch.
I absolutely love your blog.. Pleasant colors & theme. Did you build this website yourself? Please reply back as I’m planning to create my very own website and would love to find out where you got this from or exactly what the theme is called. Kudos.
кракен актуальная ссылка
Also visit my website: kraken онлайн
Смотреть серсмотреть фильмы и сериалыалы онлайн в HD — это
просто кайф!
Сериалы с русской озвучкой — идеально для фона!
зарубежные сериалы
Bununla birlikte, Milanobet bahis sitesi, kullanıcılarına hızlı ve güvenli bir ödeme deneyimi sunmak için çalışmaktadır. Ödeme işlemlerinin hızlı bir şekilde gerçekleştirilmesi için site, ödeme sürelerini minimumda tutmaktadır. Banka havalesi gibi bazı ödeme yöntemleri biraz daha uzun sürebilirken, elektronik cüzdanlar ve ön ödemeli kartlar gibi yöntemlerde ödemeler anında gerçekleşmektedir.
Зарубежные смотреть сериалы онлайн бесплатно в хорошем качестве —
мой личный топ!
Hi, I do think this is an excellent blog. I stumbledupon it 😉 I will return once again since I saved as a favorite it. Money and freedom is the greatest way to change, may you be rich and continue to guide other people.
Онлайн онлайн кинотеатр с новинками — посоветуйте лучший!
Hi there! This post could not be written much better! Going through this post reminds me of my previous roommate! He always kept talking about this. I will forward this post to him. Pretty sure he will have a very good read. Many thanks for sharing!
Great information. Lucky me I came across your website by chance (stumbleupon). I have book marked it for later!
Great site you have here.. It’s hard to find excellent writing like yours nowadays. I really appreciate people like you! Take care!!
Aproveite agora mesmo a promoção exclusiva da anchor_text para novos jogadores e comece com vantagem: ao se cadastrar gratuitamente, você recebe 100 dólares em bônus de boas-vindas. Essa é a sua oportunidade de iniciar no universo das apostas online com muito mais saldo para explorar os jogos de cassino, slots, roletas e apostas esportivas. Cadastre-se, ative sua conta e receba o bônus sem complicações. Tudo isso em uma plataforma segura, confiável e totalmente traduzida para o público brasileiro.|
punt – https://punt-us.com
bassbet – https://bassbet-de88.com
Oh my goodness! Awesome article dude! Thanks, However I am having troubles with your RSS. I don’t understand why I cannot join it. Is there anyone else having the same RSS problems? Anybody who knows the answer will you kindly respond? Thanx.
Brilliant article! https://chile-football-team.com
You need to take part in a contest for one of the best blogs on the net. I will recommend this website!
ilani casino – https://ilani-casino.com
Aproveite agora mesmo a promoção exclusiva da https://thnyjtja62.com para novos jogadores e comece com vantagem:
ao se cadastrar gratuitamente, você recebe 100 dólares em bônus de boas-vindas.
Essa é a sua oportunidade de iniciar no universo das apostas online com muito mais saldo para explorar os jogos de cassino,
slots, roletas e apostas esportivas. Cadastre-se, ative sua conta e receba
o bônus sem complicações. Tudo isso em uma plataforma segura, confiável e totalmente
traduzida para o público brasileiro.
It’s going to be finish of mine day, however before finish I am reading this fantastic article to improve my experience.|
I’m gone to say to my little brother, that he should also pay a visit this weblog on regular basis to get updated from hottest news update.|
Nice post. I was checking continuously this blog and I’m impressed!
Very helpful information specially the last part 🙂 I care for such information a lot.
I was seeking this particular information for
a long time. Thank you and best of luck.
My web blog :: anchor
I like the valuable info you provide in your articles. I will bookmark your weblog and check again here frequently. I am quite certain I’ll learn many new stuff right here! Best of luck for the next!|
rhino – https://rhino-de.com
After I originally left a comment I seem to have clicked the -Notify me when new comments are added- checkbox and from now on each time a comment is added I receive four emails with the exact same comment. Perhaps there is an easy method you can remove me from that service? Kudos.
Aproveite agora mesmo a promoção exclusiva da anchor_text para novos jogadores e comece com vantagem: ao se cadastrar gratuitamente, você recebe 100 dólares em bônus de boas-vindas. Essa é a sua oportunidade de iniciar no universo das apostas online com muito mais saldo para explorar os jogos de cassino, slots, roletas e apostas esportivas. Cadastre-se, ative sua conta e receba o bônus sem complicações. Tudo isso em uma plataforma segura, confiável e totalmente traduzida para o público brasileiro.|
We’re a group of volunteers and starting a new scheme in our community. Your web site provided us with valuable information to work on. You have done an impressive job and our entire community will be grateful to you.|
I have to thank you for the efforts you’ve put in writing this blog. I am hoping to view the same high-grade blog posts from you later on as well. In fact, your creative writing abilities has inspired me to get my own website now 😉
Hello, everything is going perfectly here and ofcourse every one is sharing information, that’s in fact fine, keep up writing.|
avaí x vila nova futebol clube – https://onde-assistir-avai-x-vila-nova-futebol-clube.com
ceará sc x brusque futebol clube – https://onde-assistir-ceara-sc-x-brusque-futebol-clube.com
Amazing issues here. I am very happy to see your post. Thank you so much and I’m taking a look forward to touch you. Will you kindly drop me a mail?|
porn
football boots – https://football-boots-eg.com
corinthians x são paulo futebol clube – https://onde-assistir-corinthians-x-sao-paulo-futebol-clube.com
Your style is unique compared to other folks I’ve read stuff from. Thank you for posting when you’ve got the opportunity, Guess I’ll just bookmark this site.
This is the right site for anyone who would like to find out about this topic. You realize so much its almost tough to argue with you (not that I really would want to…HaHa). You certainly put a fresh spin on a topic that’s been written about for years. Great stuff, just great.
Asking questions are really nice thing if you are not understanding anything fully, however this article offers good understanding even.|
seleção escocesa de futebol x seleção húngara – https://onde-assistir-selecao-escocesa-de-futebol-x-selecao-hungara.com
Howdy! This is my first visit to your blog! We are a collection of volunteers and starting a new project in a community in the same niche. Your blog provided us useful information to work on. You have done a extraordinary job!|
If you are going for most excellent contents like me, simply visit this site everyday since it presents quality contents, thanks|
Can an **Algorithmic Trading Robot for MT5 System** be used for stocks
and commodities, or just Forex?
Are there any **Forex Trading Robots** that specialize in cryptocurrency trading?
MT5 EA
From permit to final cut, Milan production service company nailed it.
Innovative service, directed event coverage in historic Italian service production company venues.
production company Milan company in Italy turned our idea into event coverage.
created in historic Italian venues beyond expectations.
Blown away!
Pro tip: book Milan production service company early. Friendly artists
books up fast! perfectly bilingual
fixer in Italy
Best video production company in hire camera crew Italy!
Reliable and Efficient, captured masterpiece in one take.
on time.
Milanobet ile sen de kazan!
Pretty portion of content. I just stumbled upon your website and in accession capital to say that I get actually enjoyed account your weblog posts.
Any way I’ll be subscribing for your augment and even I success you get admission to
consistently quickly.
Very great post. I simply stumbled upon your weblog and wished
to say that I have truly loved browsing your blog posts. After all I will be subscribing on your feed
and I am hoping you write again soon!
Howdy this is kinda of off topic but I was wanting to know if blogs use WYSIWYG
editors or if you have to manually code with HTML.
I’m starting a blog soon but have no coding skills
so I wanted to get guidance from someone with experience.
Any help would be greatly appreciated!
syria live – https://syria-live-eg.com
netbet – https://netbet-de.com
I am regular reader, how are you everybody? This paragraph posted at
this site is really pleasant.
My blog; you could try these out
I am regular reader, how are you everybody? This paragraph posted at
this site is really pleasant.
My blog; you could try these out
I am regular reader, how are you everybody? This paragraph posted at
this site is really pleasant.
My blog; you could try these out
I am regular reader, how are you everybody? This paragraph posted at
this site is really pleasant.
My blog; you could try these out
paragon casino – https://paragon-casino.com
Honkai Star Rail – https://honkaistarrail.cloud
Играю по 100 р за спин, плюсуют.
казино Эльдорадо
Играю по 100 р за спин, плюсуют.
казино Эльдорадо
Играю по 100 р за спин, плюсуют.
казино Эльдорадо
Рулетка с живым дилером — это отдельный вид кайфа.
Ставлю на красное, пью кофе, выигрываю.
казино Эльдорадо
Рулетка с живым дилером — это отдельный вид кайфа.
Ставлю на красное, пью кофе, выигрываю.
казино Эльдорадо
Рулетка с живым дилером — это отдельный вид кайфа.
Ставлю на красное, пью кофе, выигрываю.
казино Эльдорадо
Рулетка с живым дилером — это отдельный вид кайфа.
Ставлю на красное, пью кофе, выигрываю.
казино Эльдорадо
I blog quite often and I really appreciate your information. This article has really peaked my interest. I’m going to book mark your site and keep checking for new details about once a week. I opted in for your RSS feed as well.
I like reading an article that will make people think.
Also, many thanks for allowing for me to comment!
Hello, always i used to check blog posts here in the early hours in the break of day, for the reason that i love to find out more
and more.
Great information. Lucky me I recently found your website by accident (stumbleupon). I’ve saved it for later!
Джекпот в Hall of Gods — 5 млн, скрин в
чате
https://kolconexcavation.com/melbet-promokod-2025-bonusy-stavki/
Джекпот в Hall of Gods — 5 млн, скрин в
чате
https://kolconexcavation.com/melbet-promokod-2025-bonusy-stavki/
Джекпот в Hall of Gods — 5 млн, скрин в
чате
https://kolconexcavation.com/melbet-promokod-2025-bonusy-stavki/
Джекпот в Hall of Gods — 5 млн, скрин в
чате
https://kolconexcavation.com/melbet-promokod-2025-bonusy-stavki/
Играю с телевизора через браузер — большой экран
https://www.carpenteriemotta.it/2025/10/16/melbet-obzor-2025-2/
Играю с телевизора через браузер — большой экран
https://www.carpenteriemotta.it/2025/10/16/melbet-obzor-2025-2/
Бонусхантеры тут в раю, отыгровка x30 — вполне реально
https://buncitbet77.org/melbet-obzor-2025/
Бонусхантеры тут в раю, отыгровка x30 — вполне реально
https://buncitbet77.org/melbet-obzor-2025/
Бонусхантеры тут в раю, отыгровка x30 — вполне реально
https://buncitbet77.org/melbet-obzor-2025/
Бонусхантеры тут в раю, отыгровка x30 — вполне реально
https://buncitbet77.org/melbet-obzor-2025/
Бонус на выходные 50% — играю по субботам
https://bossjudi888.net/bonus-na-pervyj-depozit-melbet-2025/
Бонус на выходные 50% — играю по субботам
https://bossjudi888.net/bonus-na-pervyj-depozit-melbet-2025/
Бонус на выходные 50% — играю по субботам
https://bossjudi888.net/bonus-na-pervyj-depozit-melbet-2025/
Бонус на выходные 50% — играю по субботам
https://bossjudi888.net/bonus-na-pervyj-depozit-melbet-2025/
I’ve been surfing online more than 2 hours today, yet I never found any interesting article like yours. It’s pretty worth enough for me. In my opinion, if all web owners and bloggers made good content as you did, the internet will be much more useful than ever before.|
You should be a part of a contest for one of the greatest
blogs on the web. I will recommend this blog!
365 كورة مباشر – https://365-kora-live.com
tim nasional sepak bola jerman – https://tim-nasional-sepak-bola-jerman.com
كورا لايف 360 – https://kora-live-360.com
Ищу фулфилмент москва тарифы для маркетплейсов
в Москве, кто работает с постоплатой?
Кто может порекомендовать фулфилмент для маркетплейсов в Москве с прозрачными тарифами?
Ищу фулфилмент тарифы в Москве, который работает с Ozon и Wildberries, кто знает?
tim nasional sepak bola hong kong – https://tim-nasional-sepak-bola-hong-kong.com
бесплатные модифицированные игры — это отличный способ улучшить
игровой процесс. Особенно если вы играете
на мобильном устройстве с Android, модификации открывают перед вами большие перспективы.
Я часто использую модифицированные
версии игр, чтобы удобнее проходить игру.
Модификации игр дают невероятную свободу в
игре, что погружение в игру гораздо увлекательнее.
Играя с модификациями, я могу добавить дополнительные функции, что добавляет
новые приключения и делает игру более эксклюзивной.
Это действительно невероятно,
как такие модификации могут улучшить переживания от
игры, а при этом не нарушая использовать такие игры с изменениями можно без
особых опасностей, если быть внимательным и следить
за обновлениями. Это делает каждый игровой процесс уникальным, а возможности практически неограниченные.
Советую попробовать такие игры с модами для Android — это может добавить веселья в геймплей
Deneme bonusu, bahis siteleri arasında yeni kullanıcıların ilgisini çeken önemli bir fırsattır. Deneme bonusu veren siteler, oyunculara risksiz bir şekilde platformu tanıma imkanı sunar. Peki, bu bonusların gerçekten ne gibi avantajları ve dezavantajları var? https://lorabettvhd80.shop/
I was able to find good advice from your blog posts.
I for all time emailed this web site post page to all my associates, for the reason that if like to read it then my links will too.|
Hi, I do believe this is a great site. I stumbledupon it 😉 I may return yet again since i have book-marked it.
Money and freedom is the greatest way to change, may you be rich and continue
to help others.
Appreciate the content you put out! https://3nz9f.icu/
Хочеш зазнати успіху? найкращі онлайн казино: свіжі огляди, рейтинг майданчиків, вітальні бонуси та фрізпіни, особливості слотів та лайв-ігор. Докладно розбираємо правила та нагадуємо, що грати варто лише на вільні кошти.
Цікавлять бонуси? казино з бонусами: актуальні акції, подарунки за реєстрацію, депозитні та VIP-бонуси. Чесно розбираємо правила, допомагаємо зрозуміти вигоду та уникнути типових помилок під час гри.
Thanks for the tips! https://3nz9f.icu/man-united-had-a-howler-over-chris-smalling/
Стало легче держать спину ровной
https://storeprofit.ru/
Научился наслаждаться моментом во время
тренировки
https://storeprofit.ru/
Мульча из коры — участок как в парке
https://agro-zaschita.ru/
I found this post very helpful. https://3nz9f.icu/
Всё четко про мульчирование, сразу заказал агроволокно
https://agro-zaschita.ru/
нужна отдельная статья про скрытые расходы
https://доставка-карго-из-китая.рф/
Thanks for your marvelous posting! I truly enjoyed reading it, you happen to be a great author.I
will ensure that I bookmark your blog and definitely will
come back in the foreseeable future. I want to encourage you to ultimately continue your great job, have a
nice afternoon!
Everything is very open with a precise explanation of the challenges. It was definitely informative. Your site is extremely helpful. Thanks for sharing!|
Машина подавалась всегда чистая, без посторонних запахов
http://polyamory.wiki/index.php?title=Taxi-minvodi_97b
Fastidious response in return of this issue with real arguments and explaining all on the topic of that.
https://xn--krken23-bn4c.com
Do you mind if I quote a few of your posts as long as I provide credit and sources back to your weblog? My website is in the exact same niche as yours and my users would definitely benefit from a lot of the information you provide here. Please let me know if this alright with you. Many thanks!|
naturally like your website but you need to check the spelling on quite a few of your posts. Many of them are rife with spelling problems and I to find it very troublesome to tell the reality however I will certainly come again again.|
химчистка обуви из замши химчистка обуви
Valuable information. Fortunate me I discovered your site by chance, and I’m stunned why this twist of fate did not came about earlier! I bookmarked it.
Loved the insight provided in this post. It’s very well-written and packed with valuable details. Fantastic job!
Really liked it! Recommend.
Excellent pieces. Keep posting such kind of info on your site. Im really impressed by your site.
This article is fantastic. I learned a lot from going through it. The information is very informative and arranged.
I have been browsing online more than three hours today, yet I never found any interesting article like yours. It’s pretty worth enough for me. In my opinion, if all webmasters and bloggers made good content as you did, the net will be much more useful than ever before.
thanks for sharing
Great work! This is the type of info that should be shared around the web. Shame on the search engines for not positioning this post higher! Come on over and visit my site . Thanks =)
Great packaging. Loved it.
интернет магазин люстр люстра светильник деревянный
бренды мужских костюмов мужской костюм купить магазин
This post is fantastic. I gained tons from going through it. The content is highly educational and arranged.
Appreciated the information in this article. It’s extremely well-researched and full of useful insights. Excellent work!
WOW just what I was searching for. Came here by searching for b2xbet
Great product! Really impressed with the quality.
18bet – https://18betmx.com
It’s really a great and helpful piece of information. I am satisfied
that you just shared this useful info with us. Please keep us informed like this.
Thanks for sharing.
I love your blog.. very nice colors & theme.
Did you create this website yourself or did you hire someone to do
it for you? Plz answer back as I’m looking to create my own blog and would like to
know where u got this from. thank you
10bet – https://www-10bet-mx.com
Franchement, je remarque que le secteur du iGaming se transforme vraiment rapidement en ce moment.
L’autre jour, j’ai essayé quelques nouvelles interfaces
et je constate que l’ergonomie est généralement bien plus soigné qu’avant.
Par exemple, en analysant les possibilités de spino gambino casino, on réalise directement que la sécurité des transactions devient le facteur
majeur pour les parieurs. De mon point de vue, jouer petit sur des cotes stables est souvent la meilleure méthode pour
tenir dans la session. Aussi, j’ai vu que le SAV réagit plus vite tard, c’est assez curieux comme détail.
Est-ce que un membre ici a récemment utilisé une application nomade qui demande vraiment le détour?
Je serais vraiment ravi d’en parler ici même. Quels sont vos avis concernant cette nouvelle?
I like the helpful info you provide to your articles. I’ll bookmark your weblog and test again right here regularly. I’m slightly sure I’ll be informed plenty of new stuff proper here! Best of luck for the next!|
Ресурсы вроде взломанные игры на андроид предоставляют крупный список взломанных игр на андроид, поддерживая полную среду вокруг взломов.
Такой тренд сформировала сообщество пользователей, которые стремятся получить улучшенные
возможности без вложений.
It’s very trouble-free to find out any topic on net as compared to
textbooks, as I found this paragraph at this website.
Пользовались услугами таможенного брокера
в Москве — оформили груз быстро и без лишних вопросов,
остались довольны
Here is my homepage … таможенное оформление москва
Excellent article. I found the content highly helpful. Appreciated the method you detailed all the points.
I read this post fully about the comparison of most up-to-date and preceding technologies,
it’s remarkable article.
I believe that is one of the so much significant info for me.
And i’m satisfied studying your article. But want to statement on some general things, The website taste is ideal, the articles is in reality nice : D.
Just right task, cheers
Таможенные услуги Москва рекомендую для импорта и экспорта
Check out my site: таможенное сопровождение москва
Great weblog here! Additionally your website a lot up very fast! What host are you using? Can I am getting your associate link in your host? I desire my web site loaded up as quickly as yours lol
I was just looking for this information for some time. After 6 hours of continuous Googleing, at last I got it in your web site. I wonder what is the lack of Google strategy that do not rank this type of informative websites in top of the list. Generally the top web sites are full of garbage.
Tüm maçları canlı yayınla izlemek için siz de Tarabet TV’ye gelin.
Sweet bonanza’da güvenilir siteleri arıyorsanız sizde sitemizi ziyaret edebilirsiniz.
Решили вопрос с запретом на ввоз из параллельного импорта
Here is my blog post: таможенный представитель москва
Everything perfect. Very well-made.
аккумулятор на электропогрузчик купить Батарея тяговая для Komatsu — емкость 500-800 Ач. Полная совместимость.
Ремонт деревянного покрытия Реставрация картины
Hey this is somewhat of off topic but I was wanting to know if blogs use WYSIWYG editors
or if you have to manually code with HTML. I’m starting a blog soon but have no coding know-how so
I wanted to get guidance from someone with experience. Any help would
be enormously appreciated!
Disability Rights
codigo promocional 1xbet venezuela 2026
company set up uae whitecirclegroup company setup
Купить москитные сетки
топливо купить
строительные леса аренда
Аренда манипулятора
скупка золота грамм
продвижение сайтов в Санкт-Петербурге
раскрутка сайтов москва
https://sozdatelisaitov.ru/
Футбольные ставки Football Predictions with expert tipsters on betting tips, Fixed game and Correct Score Best exacte Forecast Sports Games Matches Bets VIP Free Daily Today Sureshot Odds Accumulator Real 100%
Установка люстры Севастополь Читайте отзывы об услугах электрика в Севастополе и убедитесь в нашей квалификации.
переборка тормозных суппортов Наши мастера проводят замену тормозных дисков, восстанавливая эффективность всей системы.
https://spisok-kreditov.ru/ На doskazaymov можно подобрать кредит наличными без лишних вопросов.
Футбольные ставки Expert tipsters deliver betting tips on sports games and matches including Fixed game selections and correct score best exacte forecast. Vip and free bets are available daily with sureshot odds and accumulator real
ко ланта таиланд
I’ve read several excellent stuff here. Definitely worth bookmarking for revisiting.
I surprise how a lot effort you set to create one of these wonderful informative website.
My developer is trying to persuade me to move to .net from PHP. I have always disliked the idea because of the expenses. But he’s tryiong none the less. I’ve been using Movable-type on a variety of websites for about a year and am concerned about switching to another platform. I have heard fantastic things about blogengine.net. Is there a way I can transfer all my wordpress content into it? Any kind of help would be really appreciated!|
It’s hard to come by well-informed people for this topic, but you seem like you know what you’re talking about! Thanks|
кайт сафари Египет Кайт кайтинг кайтсёрфинг школа обучение сафари Дети ветра DETIVETRA
Have you ever considered writing an e-book or guest authoring on other websites? I have a blog based upon on the same subjects you discuss and would love to have you share some stories/information. I know my viewers would enjoy your work. If you are even remotely interested, feel free to send me an e-mail.|
Hi i am kavin, its my first occasion to commenting anyplace, when i read this paragraph i thought i
could also make comment due to this good piece of writing.
школы кайтсерфинга Хургада Кайт кайтинг кайтсёрфинг школа обучение сафари Дети ветра DETIVETRA
Hello There. I found your weblog the use of msn. That is a very well written article. I will make sure to bookmark it and return to read more of your useful info. Thank you for the post. I will definitely return.|
дизайн интерьера 3д бюро дизайна интерьера
Der Ursprung von chicken road seriös hat ein außergewöhnliches Basis, da das
chickenroad game ursprünglich als humorvolle Idee entstand, bei der ein winziges Huhn gefährliche Routen passiert und
dabei vielfältige Hindernisse bewältigt. Im Laufe der Zeit verwandelte sich
chicken road zu einem Spielkonzept, der sowohl Gamer als
auch Entwicklern prägnant haftengeblieben ist, weil er ein einfaches aber fesselndes Grundkonzept verkörpert,
das leicht verständlich, aber gleichzeitig fordernd ist.
Mit wachsender Popularität entstand auch die mobile chickenroad-Version, welche vielen Anwendern ein flexibles Spielgefühl bot und das
Konzept in kurzer Zeit verbreitete. Während manche Spieler chicken road erfahrungen als
nostalgisch empfinden, betonen andere die Fähigkeit des Games, moderne Mechaniken mit traditionellem Spielgefühl
zu kombinieren, was die Basis dafür wurde, dass Chicken Road heute auch in anderen Varianten genutzt wird.
Die Spieleentwickler erweiterten das Konzept und
schufen Varianten wie Chicken Road Demo für Anfänger und Chicken Road 2 als zweiten Teil, die eine lebendigere
Game-Welt und zusätzliche Herausforderungen einführte.
Auffällig ist, dass das chicken road casino in einigen Versionen als Designmotiv auftauchte, bei dem Abläufe
an spielhallenähnliche Strukturen erinnerten, ohne jedoch den Kern des Spiels zu verlieren. Da viele Spieler Wert darauf legten zu wissen, ob chickenroad seriös sei, versuchten die Entwickler stets klar zu arbeiten und ein gerechtes Spielsystem zu bieten, was das
Vertrauen der Community über Jahre hinweg stärkte.
Diese kontinuierliche Evolution, kombiniert mit kreativen Anpassungen, sorgte dafür,
dass chickenroad Slot Elemente in einigen Editionen integriert wurden, wodurch
das Spielerlebnis erweitert wurde und neue Nutzergruppen erreicht werden konnten.
Letztlich zeigt die Geschichte von chickenroad, wie ein einfaches Konzept durch beständige Weiterentwicklung,
Anpassungsfähigkeit und eine motivierte Nutzergemeinschaft wachsen kann und zu einem breiten Spielekosmos wird, das diverse Nutzer anspricht und in verschiedenen Formen fortbesteht.
Thanks for the good writeup. It if truth be told was once a enjoyment account it. Look complicated to more introduced agreeable from you! By the way, how can we be in contact?|
WOW just what I was looking for. Came here by searching for
gbgbet
Its like you read my mind! You seem to know a lot about this, like you wrote the book in it or something. I think that you can do with some pics to drive the message home a little bit, but instead of that, this is great blog. An excellent read. I will certainly be back.|
Oh my goodness! Impressive article dude! Thank you, However I am experiencing problems with your RSS. I don’t know the reason why I can’t join it. Is there anybody having the same RSS problems? Anyone who knows the solution can you kindly respond? Thanx!!|
Howdy! This article couldn’t be written much better! Looking through this post reminds me of my previous roommate! He continually kept talking about this. I am going to forward this article to him. Fairly certain he’ll have a very good read. Thanks for sharing!|
1xbet mobil yükle çox rahatdır, hər kəsə məsləhət görürəm
1xbet indir android
https://auto.ae/catalog/
https://auto.ae/showrooms/all/
арендовать минивэн мерседес по часам mercedes vito аренда с водителем
sparkdex ai SparkDex is redefining decentralized trading with speed, security, and real earning potential. On spark dex, you keep full control of your assets while enjoying fast swaps and low fees. Powered by sparkdex ai, the platform delivers smarter insights and optimized performance for confident decision-making. Trade, earn from liquidity, and grow your crypto portfolio with sparkdex — the future of DeFi starts here.
Фармилки Каждая из этих сфер – майнинг, аирдропы, тапалки, фармилки – предлагает свой уникальный путь к заработку криптовалюты, а интеграция с Telegram делает их доступными каждому.
https://emiratesambassadeurs.com/vip-concierge/ Zurich: EA Private Desk in Geneva can check availability for Richard Mille RM 67-01 and arrange Ferrari Group insured delivery before invoicing.
I’m gone to say to my little brother, that he should also visit this website on regular basis to take updated from latest information.|
проверка прокуратуры Волгоград Добро пожаловать на новостной портал Волгограда. Мы публикуем последние и главные новости региона, происшествия и криминальную хронику.
bs2we at
http://ekaraganda.kz/?mod=news_read&id=120522&site-admin=1
кайтинг кайтсёрфинг
https://japansan.ru/images/art/?1xbet_promokod_pri_registracii_bonus_segodnya.html
bs2we at
лазерные принтеры (Лазерный принтер – идеальное решение для быстрой и четкой печати документов. | Лазерные принтеры превосходят струйные по скорости и экономии тонера. | Хотите лазерный принтер купить? Широкий выбор моделей по доступным ценам! | Лазерные принтеры купить легко в нашем магазине с гарантией качества. | Купить лазерный принтер – значит инвестировать в надежность и производительность. | Заказать лазерный принтер онлайн – быстро и без лишних хлопот. | Лазерный принтер цена радует: от 5000 руб. за базовые модели. | Узнайте лазерный принтер стоимость – выгодные акции для всех покупателей. | Ищете лазерный принтер недорого? У нас лучшие предложения! | Лазерный принтер купить недорого – реальность с нашими скидками до 30%. | Дешевый лазерный принтер не уступает по качеству печати. | Бюджетный лазерный принтер для дома и офиса – оптимальный выбор. | Лазерный принтер купить онлайн в 2 клика с доставкой. | Заказать лазерный принтер онлайн – удобный сервис 24/7. | Лазерный принтер интернет магазин с тысячами отзывов. | Интернет магазин лазерных принтеров – ваш надежный партнер. | Лазерный принтер каталог: фото, характеристики, отзывы. | Лазерный принтер в наличии – забирайте сегодня! | Лазерный принтер с доставкой по России бесплатно от 5000 руб.)
сатира про бюрократию жизнь в россии без прикрас
заказать аудиоролик Заказать аудиорекламу просто: мы создадим яркий ролик с учетом вашего бренда.
No matter if some one searches for his vital thing, therefore he/she wishes to be available that
in detail, so that thing is maintained over here.
Hurrah! At last I got a weblog from where I know how to truly obtain useful data concerning my study and knowledge.|
Appreciated this entry. It’s extremely well-researched and filled with helpful insights. Excellent job!
blsp at
тарифы +в мегафон Выбор подходящего тарифа интернета от Мегафон или другого провайдера важен для комфортного пользования услугами, будь то для работы, развлечений или телевидения.
https://www.instagram.com/seo_s_umom_by/ продвижение сайтов
https://www.studenternas.nu
https://pastelink.net/1csfjq60
bs2best at blsp at
трансы воронеж Каждый трек — это путешествие, построенное на постепенном нарастании напряжения и последующем катарсисе, приводящем к взрыву эмоций.
ко ланта как добраться ко ланта таиланд
http://firstrainingsalud.edu.pe/profile/BonusXaf6/
Hi there, You’ve done a great job. I’ll definitely digg it and personally recommend to my friends. I am sure they will be benefited from this website.|
свирыч SWeAR2033 умеет создавать пространство, где каждый чувствует себя комфортно и вовлеченно, что и является залогом успеха его стримов.
кухни на заказ в Москве Наша цель — показать, что «алгоритмы» могут быть не только функциональными, но и элегантными, «вкусными» и захватывающими.
Can I simply just say what a comfort to uncover someone who genuinely knows what they are discussing on the web. You certainly understand how to bring an issue to light and make it important. More and more people have to read this and understand this side of the story. I was surprised that you’re not more popular since you surely have the gift.|
кайтсёрфинг кайтсёрфинг
забор звенигород
https://vrwant.org/wb/home.php?mod=space&uid=4967755
http://www.lvovvv.ru/user/gwaniereum
расписание автобусов из луганска до москвы
Нужны столбики? столбики для ограждения с лентами столбики для складов, парковок и общественных пространств. Прочные материалы, устойчивое основание и удобство перемещения обеспечивают безопасность и порядок.
свадебное платье рыбка каталог свадебных платьев с ценами
купить кабель 6 проводка цена
Looking for a yacht? family boat rental Cyprus for unforgettable sea adventures. Charter luxury yachts, catamarans, or motorboats with or without crew. Explore crystal-clear waters, secluded bays, and iconic coastal locations in first-class comfort onboard.
кайт школа в крыму кайтинг
аирдроп криптовалют Крипта, майнинг, аирдропы и тапалки — всё самое свежее в Smart Money Crypto Зарабатывай на криптоиграх и фармилках прямо в Telegram. Пассивный доход с удовольствием и без вложений! Заходи и забирай свою прибыль. ИЛИ Ищешь где заработать на крипте? Smart Money Crypto Новости, криптоигры с выводом, актуальные аирдропы и майнинг на телефоне. Тапалки и фармилки в Telegram для твоего пассивного дохода. Присоединяйся к команде Smart Money!
https://sites.suffolk.edu/pierpaolo19/2012/10/28/mit-nuclear-reactor-tour/#comment-248463
Профильная труба купить в Алматы
casino mafia Decouvrez Mafia Casino avis 2026 : site officiel avec connexion securisee, bonus exclusifs, milliers de jeux et retraits rapides. Inscrivez-vous des maintenant!
https://minecraftreward.com/code-promo-1xbet-gratuit-aujourdhui-bonus-100/
hello there and thank you for your info – I’ve certainly picked up something new from right here. I did however expertise several technical points using this site, since I experienced to reload the website many times previous to I could get it to load properly. I had been wondering if your web host is OK? Not that I am complaining, but sluggish loading instances times will often affect your placement in google and can damage your quality score if advertising and marketing with Adwords. Anyway I’m adding this RSS to my email and could look out for much more of your respective interesting content. Ensure that you update this again very soon.|
Highly descriptive post, I liked that bit. Will there be a part 2?
We are a group of volunteers and opening a new scheme in our community.
Your website offered us with useful information to work
on. You’ve done an impressive activity and our entire community might be thankful
to you.
размеры металлических значков изготовил металлических значков москва
кинг казино официальный Ребята, искал вообще рабочее зеркало для кинг казино и случайно вышел на этот канал. Там выкладывают актуальные ссылки почти каждый день, плюс бонусы иногда дают. Я уже пару раз пользовался, все открывается без проблем. Не заметил никаких глюков или косяков. Думаю, можно смело юзать
Do you mind if I quote a couple of your articles as long as I provide credit and sources back to your website? My website is in the very same niche as yours and my visitors would genuinely benefit from a lot of the information you present here. Please let me know if this okay with you. Many thanks!|
ауф казино зеркало
Ребята, наткнулся на этот канал по ауф казино, решил закинуть сюда, потому что тема реально полезная. Там регулярно выкладывают актуальные ссылки для входа, плюс иногда появляются бонусы и промокоды. Я уже несколько раз заходил через них — все открывается без проблем, без странных редиректов и лишних действий. В целом выглядит как норм рабочий вариант, можно сохранить на случай если основной сайт не открывается
Kraken Marketplace Официальный сайт для доступа к Kraken
krab cc
Kraken Marketplace Официальный сайт для доступа к Kraken
kraken
Kraken Marketplace Официальный сайт для доступа к Kraken
krab cc
Kraken Marketplace Официальный сайт для доступа к Kraken
kra at
https://t.me/tigercasinoofficial/14
Хотел поиграть без заморочек, tiger casino норм
Kraken Marketplace Официальный сайт для доступа к Kraken
slon at
Kraken Marketplace Официальный сайт для доступа к Kraken
kraken market
Kraken Marketplace Официальный сайт для доступа к Kraken
slon4 at
Kraken Marketplace Официальный сайт для доступа к Kraken
krab at
Kraken Marketplace Официальный сайт для доступа к Kraken
slon at
Kraken Marketplace Официальный сайт для доступа к Kraken
kraken
Kraken Marketplace Официальный сайт для доступа к Kraken
slon4 at
Kraken Marketplace Официальный сайт для доступа к Kraken
slon at
рабочее зеркало eva casino
Искал eva casino где войти, нашел тут
This design is incredible! You definitely know how to keep a
reader amused. Between your wit and your videos, I
was almost moved to start my own blog (well, almost…HaHa!) Fantastic job.
I really loved what you had to say, and more than that, how you presented it.
Too cool!
Hey I know this is off topic but I was wondering if you knew of any widgets I could add to my blog that automatically tweet my newest twitter updates. I’ve been looking for a plug-in like this for quite some time and was hoping maybe you would have some experience with something like this. Please let me know if you run into anything. I truly enjoy reading your blog and I look forward to your new updates.|
Sweet bonanza oynamak için güvenilir slot siteleri arıyorsan doğru adrestesin.
декоративная цепочка с крестом
Нашел интересную серебристую подвеску с крестом для альтернативных образов. Подойдет тем, кто любит gothic, opium и y2k эстетику
Андрей Аршавин Telegram
Нашел телеграм Аршавина, все работает
Матвей Сафонов официальный telegram
Искал, нашел
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blsp сайт
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
bs2best вход
https://t.me/fenixcasino_official
Парни, если кто ищет fenix casino вход или рабочее зеркало, вот норм вариант через телеграм. Канал живой, обновляется часто, выкладывают актуальные ссылки и бонусы. Я сначала сомневался, потому что сейчас много фейков, но проверил — все реально открывается. Уже пару раз пользовался, проблем не заметил
Bayilik ve yeni iş fikirleri konularında içerikler okumak ve kendini geliştirmek istiyorsan senin de adresin https://baygirisimci.com/ olsun
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
bs2best at
Este ambiente de casas de aposta digitais vem crescido com velocidade, e
sites tipo o high fly bet casino são reconhecidos por oferecer
opções para apostas inovadoras e imersivas. Esse high fly
bet, como é chamado, une a tecnologia estrutura avançada e interface
estrutura simples, permitindo uma jornada clara para os usuários novatos como também para profissionais.
Um dos pontos fortes do casino highflybet é a
variedade dos opções disponíveis, os quais inclui caça-níqueis populares e também games de
mesa complexos, tais como pôquer junto com roleta ou pôquer.
Essa mesma opção propicia que os usuários encontrem sempre a alternativa que encaixe ao seu estilo de diversão
e às suas escolhas pessoais. Além do mais, o high flybet aplica
de forma contínua em segurança online, por meio de criptografia sofisticada a fim de garantir informações pessoais bem como monetários dos
usuários. Além disso, fator que ajuda ao crescimento da casino
highflybet consiste em o suporte ao cliente, que apresenta eficiente e prático, atendendo questões e oferecendo assistência de modo ágil bem como direta.
Com a fama em crescimento, este casino high fly bet vem atraindo a credibilidade do seu usuários em expansão, estabelecendo-se como uma referência segura no setor cassinos virtuais.
Através de ofertas, prêmios e clubes de pontos, o serviço pode garantir usuários ativos, incentivando a experimentação de diversos apostas bem como melhorando a satisfação total dos
usuários. Portanto, este high fly bet além de oferece
diversão, mas também oferece proteção, diversidade junto com tranquilidade, elementos importantes de quem procura experiência plena
dentro do ambiente de jogos online.
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blacksprut marketplace
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blacksprut
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
bs2best вход
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
bs2best at
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blsp at
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blsp сайт
wonderful post, very informative. I’m wondering why the other specialists of this sector do not notice this. You should proceed your writing. I’m confident, you’ve a great readers’ base already!|
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
bs2best at
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blsp at
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
bs2best вход
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blacksprut
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blacksprut marketplace
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
bs2best вход
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blacksprut
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blsp сайт
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
bs2web at
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
bs2best at
ева казино
Не идеал но рабочий
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blsp сайт
most bet se postupně získal jedním z nej z nejpopulárnějších brandů v sektoru digitálního sázení plus hazardních
platform zatímco její působení se neomezuje limitováno výhradně
v určité území, jelikož, nabídky mostbet download poskytují velkou škálu možností sázkařům v celé EU.
Vznik mostbet se charakterizuje snahou spojit běžné principy sázení za pomoci moderními technologiemi, a tím umožnilo stránce rychle dosáhnout uznání u profesionálními sázkaři i nové uživatele.
mostbet casino poskytuje také sázkové sázky, a také také tradiční herní hry, a to přitahuje rozsáhlou veřejnost.
Díky tomuto tradici a stabilnímu chodu se most bet vybudoval
důvěru solidního hráče v online sázení.
Společnost by se soustreďuje na otevřenost, bezpečnost a
férové zásady, které představuje zásadní ohledně posílení stálé věrnosti uživatelů.
Protože, že Mostbet app poskytuje rychlý vstup k sázení a hře přímo přes přenosného zařízení, uživatelé mohou
využívat funkce po celý čas i všude, aniž to byli omezeni počítačem.
Funkce stahování app skrze Most Bet download umožňuje, aby celá aspekty jsou optimalizované
pro rychlý plus snadný zážitek. Také faktorem, který
přispívá pro popularitě mostbet, znamená široká paleta bonusů, co motivují nové
i pravidelné sázkaře, ať už se vztahuje o uvítací mostbet bonus
či pravidelné nabídky v rámci herního prostředí. Dějiny Mostbet prokazuje,
že platforma byla schopna efektivně adaptovat se ohledně
požadavky obchodu, přizpůsobovat volby plus modernizovat, a tím garantuje platformy stabilní reputaci v rámci konkurencí.
Souhrnně lze prohlásit, že platforma pevné základy, sloučení klasického a pokročilého metody
a intenzivní zaměření na uživatelský skúsenost znamená střet úspěšnosti
most bet a jeho možností například Most Bet CZ nebo Most Bet Casino, a tím z něj oblíbenou variantu pro velkou cílovou skupinu hráčů.
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blacksprut marketplace
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blsp at
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blacksprut marketplace
Paragraph writing is also a fun, if you be acquainted
with after that you can write otherwise it is complicated to
write.
I blog quite often and I truly thank you for your content. This great article has truly peaked my interest. I will take a note of your site and keep checking for new information about once a week. I subscribed to your Feed too.|
Die Entstehung von chickenroad trägt ein beeindruckendes Konzept, da das chicken road game anfänglich als witzige Eingebung
entstand, bei der ein winziges Huhn gefährliche Straßen überquert und dabei verschiedene
Blockaden bewältigt. Mit der Zeit verwandelte sich chicken road zu einem Namen, der sowohl Nutzern als
auch Entwicklern einprägsam haftengeblieben ist, weil er ein geradliniges aber packendes Grundkonzept darstellt, das einfach
nachvollziehbar, aber gleichzeitig herausfordernd ist.
Aufgrund steigender Bekanntheit entstand auch
die mobile chickenroad-Version, welche vielen Anwendern ein unterwegs verfügbares Spiel bot und das Konzept in kurzer Zeit verbreitete.
Während manche Spieler Erfahrungen mit Chicken Road als
klassisch empfinden, betonen andere die Stärke des Spiels, moderne
Systeme mit klassischem Gameplay zu verbinden, was die Basis dafür wurde,
dass chickenroad heute auch in anderen Varianten genutzt wird.
Die Spieleentwickler erweiterten das Design und schufen Varianten wie chickenroad Demo für Anfänger
und Chicken Road 2 als zweiten Teil, die eine dynamischere Game-Welt und zusätzliche Herausforderungen einführte.
Bemerkenswert ist, dass das Chicken Road Casino in einigen Versionen als Designmotiv auftauchte, bei dem
Spielmechaniken an Glücksspiele erinnerten, ohne jedoch den eigentlichen Charakter des Spiels zu verlieren. Da viele Spieler Wert darauf legten zu wissen, ob chickenroad seriös sei, versuchten die Entwickler stets transparent
zu arbeiten und ein gerechtes Spielsystem zu bieten, was
das Vertrauen der Nutzerbasis über Jahre hinweg stabilisierte.
Diese laufende Anpassung, kombiniert mit gestalterischen Erweiterungen, sorgte dafür, dass Chicken Road
Slot Elemente in einigen Editionen integriert wurden, wodurch das Gameplay
erweitert wurde und neue Spielertypen erreicht werden konnten. Letztlich zeigt die
Geschichte von chicken road, wie ein einfaches Konzept durch
kontinuierliche Pflege, Flexibilität und eine aktive
Spielerschaft wachsen kann und zu einem vielseitigen Universum wird, das verschiedene Player anspricht und in verschiedenen Formen fortbesteht.
office space for rent new york https://offices-rent-nyc.com
Sweet blog! I found it while surfing around on Yahoo News. Do you have any tips on how to get listed in Yahoo News? I’ve been trying for a while but I never seem to get there! Cheers|
Piece of writing writing is also a fun, if you know after that you can write otherwise it is complex to write.|
ева казино вход
Если нужен доступ к ева казино, через телеграм это самый простой вариант
Het spel Plinko blijft het onder populairste meest gewilde casinospellen dat online
en internet als offline wordt gespeeld, terwijl plinko casino blijft
zichzelf bewezen als het eenvoudig maar interessant game voor
gebruikers met alle levels. Het principe van Plinko is gemakkelijk voor snappen, en dat helpt tot de interesse daarin: een gebruiker gooit de bal en disc
vallen op het rand van een vak, waar gevuld blijkt van pins dan blokken, doch het schijf rolt random naar laag totdat de bal op
een onder de vakken onderaan terechtkomt,
ieder slot bevat het specifieke waarde of verdienste. Het Plinko uniek zorgt,
vooral in de digitale omgeving zoals in een Plinko platform, blijft de mix met eenvoud met
sensatie; spelers hoeven niet moeilijke plannen te snappen, en toch alle gooi biedt het sensatie voor spanning plus sensatie.
Binnen NL groeit het populariteit van Plinko spel flink, zeker op sites die gerenommeerd worden als NL Plinko, daar gebruikers makkelijk inlog verkrijgen voor proefversies
plus geld betaalversies op Plinko gokspel. Aan beginners is het vraagstuk “hoe werkt Plinko?” vaak het eerste actie in de verkenning, want een regels eenvoudig blijven maar
de uitkomsten wisselend en ook random. Daarnaast blijkt het spel spel regelmatig gelauwerd bij reviews Plinko om het zichtbare interesse en ook de simpele
om te volgen systeem die plus casual spelers plus hardcore casino bezoekers trekt.
Het online internet game Plinko spel biedt daarnaast regelmatig
extra opties bijvoorbeeld inzetopties, multipliers en speciale rondes dat
de game dynamischer brengen, zodat deelnemers hun ervaringen willen bijstellen op grondslag voor hun instellingen plus risicobereidheid.
Blijkt Plinko spel inderdaad eerlijk plus geloofwaardig?
Veel gebruikers vragen hun dat, en ook betrouwbare casino’s tonen de
spelmechanismen plus payouts duidelijk, waardoor verbetert aan het
waardering voor Plinko veilig binnen de casino gemeenschap.
Concluderend, Plinko voegt samen een eenvoudige structuur en spannende scores en ook beschikbaarheid, zodat het het onmisbaar onderdeel is van voor het actuele digitale casino ervaring.
официальный пост eva casino
Норм вариант без лишнего мусора
феникс казино тг
Сначала не получалось зайти вообще, потом подсказали что через тг канал проще. В итоге реально помогло, перешел по ссылке и все заработало
With havin so much content and articles do you ever run into any issues of plagorism or copyright infringement? My site has a lot of completely unique content I’ve either created myself or outsourced but it seems a lot of it is popping it up all over the web without my agreement. Do you know any ways to help stop content from being ripped off? I’d truly appreciate it.|
Pretty component of content. I just stumbled upon your site and in accession capital to say that I acquire actually enjoyed account your weblog posts. Any way I’ll be subscribing to your augment or even I achievement you get right of entry to persistently quickly.|
This site was… how do you say it? Relevant!! Finally I’ve
found something which helped me. Thanks!
Приятно удивлен качеством упаковки, паллеты пришли как новенькие.
Here is my website :: Сборные грузы из Китая в Россию
It’s really a cool and useful piece of information. I am satisfied that you shared this useful information with us.
Please stay us informed like this. Thank you for sharing.
Excellent blog! Do you have any recommendations for aspiring writers? I’m planning to start my own blog soon but I’m a little lost on everything. Would you suggest starting with a free platform like WordPress or go for a paid option? There are so many choices out there that I’m completely confused .. Any recommendations? Many thanks!|
I blog frequently and I seriously appreciate your information. Your article has truly peaked my interest. I’m going to take a note of your blog and keep checking for new details about once a week. I subscribed to your RSS feed too.|
https://t.me/s/fenixcasino_official
Через него сейчас и захожу
Hi there! I could have sworn I’ve been to this web site before but after browsing through some of the articles I realized it’s new to me. Anyways, I’m certainly pleased I stumbled upon it and I’ll be book-marking it and checking back frequently!|
Genuinely no matter if someone doesn’t understand then its up to other visitors that they will help, so here it occurs.|
Wrapped up a ripper night session punting on crash games and have to say nothing beats the rush when you work out the right time to cash out. Gave a crack https://rootskitchen.bar last night and was stoked. Funded my account with Visa and saw they also have Skrill which suits Aussies well. The multiplier climb is genuinely exciting — you gotta hold your nerve while also not get greedy. Compared to Lucky Jet, Aviator which I’ve also punted on, it offers a genuine live split-second thrill. Good setup for Aussie players — worth a visit at https://rootskitchen.bar
Este universo de chickenroad ha evolucionado con velocidad desde su aparición inicial como una idea
sencilla, ligera y dirigida a quienes buscaban un juego dinámico y
ligero. Después, frases como juego chicken road o hasta chicken road 2 empezaron a aparecer en charlas de jugadores que querían entender si esta
experiencia ofrecía diversión, si incluía desafíos y si mantenía el interés.
Al mismo tiempo, aparecieron preguntas sobre si chicken road es real, opiniones chicken road y
el interés creciente sobre casino chicken road, lo que convirtió al juego
en un fenómeno lúdico. En este primer apartado conviene ver
por qué chckenroad captó tanta atención: su forma funciona avanzando, esquivar obstáculos y mantener la velocidad, una fórmula común por muchos títulos, aunque aquí
se une con una estética ligera y un ritmo rápido que empuja a seguir.
Estos sistemas han dado lugar charlas en comunidades digitales
sobre si es solo casual o si entra en un segmento mayor
donde hay dinámicas competitivas. También, los comentarios
chicken road reflejan que muchos jugadores vieron equilibrio entre diversión y tensión, lo que ayudó su difusión en territorios de habla hispana.
La gente revisan cómo chicken road 2 ajustó la experiencia con más niveles, movimiento más suave
y una imagen renovada. Sin embargo, algunas charlas tocan el
tema de si realmente aporta algo nuevo o solo mejora la fórmula inicial.
La llegada del término casino chicken road
surge por la curiosidad de quienes quieren saber si tiene vínculo con azar, lo
que creó debates, pero en esencia chicken road sigue
apoyado en coordinación. Todo este conjunto ha vuelto a chicken road una referencia dentro de
los juegos casuales, donde la simplicidad es su mayor atractivo.
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
bs2 market
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blsp at
These are really impressive ideas in regarding blogging. You have touched some pleasant factors here. Any way keep up wrinting.|
Hi! I’m at work surfing around your blog from my new iphone 4! Just wanted to say I love reading your blog and look forward to all your posts! Carry on the fantastic work!|
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blsp сайт
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blsp сайт
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
bs2 market
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
bs2 market
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blacksprut
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
bs2 market
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blsp сайт
eva casino login
Вообще сейчас с eva casino есть проблема, что куча клонов в выдаче, поэтому лучше сразу идти через официальный телеграм, там есть норм ссылка
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blsp сайт
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blacksprut
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
blacksprut marketplace
BlackSprut marketplace официальный сайт для входа и регистрации
blsp at bs2best at
bs2 market
I don’t know whether it’s just me or if everybody else experiencing issues with your blog. It appears like some of the written text within your content are running off the screen. Can somebody else please provide feedback and let me know if this is happening to them as well? This could be a problem with my internet browser because I’ve had this happen before. Cheers|
This article is genuinely a nice one it helps new web people, who are wishing for blogging.|
play jonny, oft unter anderem bekannt als casino playjonny genannt, wurde
in den vergangenen Jahren als eine eine bedeutende bemerkenswertesten Anbieter im Sektor Online-Casinos etabliert.
Diese Plattform verbindet fortschrittliche Technologie und
einer benutzerfreundlichen Oberfläche, die für für Anfänger als auch für erfahrene Spieler leicht bedienbar bleibt.
Besonders hervorzuheben bleibt das Angebot der Spiele, die von klassischen Spielautomaten über Tischgame und
auch Live-Erfahrungen angeboten werden. Diese vielfältige Auswahl macht playjonny für einer attraktiven Option für Gamer, die Unterhaltung und und auch die Chance auf Gewinne suchen. Zusätzlich achtet play jonny casino starken Fokus auf Datenschutz und Sicherheit, wodurch Spieler
ihre persönlichen Daten Transaktionen ohne Sorgen abwickeln können. Ein weiterer Vorteil des
PlayJonny Casinos zeigt sich das regelmäßige Update des Spieleangebots, wodurch Gamer
ständig neue Optionen finden können. Zusätzlich zur
Auswahl der Spielangebote überzeugt play jonny ebenfalls mit innovativen Funktionen, wie Bonussystemen,
Treuepunkten und regelmäßigen Aktionen, welche das Spielerlebnis interessanter verbessern. Insgesamt zeigt sich, dass nämlich play jonny nicht nur ein gewöhnliches Online-Casino besteht, sondern vielmehr eine
Plattform, die für die Anforderungen moderner Nutzer ausgerichtet ist und dabei Unterhaltung
und Fairness bietet. Die Kombination aus Benutzerfreundlichkeit, Vielfalt und Sicherheit
gestaltet play jonny casino zu einem herausragenden Akteur im Online-Glücksspielmarkt.
Официальная ссылка маркетплейса “Зелёный Мир”
маркетплейс
Официальная ссылка маркетплейса “Зелёный Мир”
Любители каннабиса
Официальная ссылка маркетплейса “Зелёный Мир”
Зелёный Мир
Официальная ссылка маркетплейса “Зелёный Мир”
Зелёный_Мир
It’s hard to find experienced people about this subject, however, you seem like you know what you’re talking about!
Thanks
Официальная ссылка маркетплейса “Зелёный Мир”
3мир магазин
изготовить флаг на заказ изготовление флагов
Do you want to go to Montenegro? https://www.holidays-in-montenegro.com an Adriatic holiday with pristine beaches and beautiful cities. Resorts, excursions, and active recreation. An ideal destination for travel and seaside relaxation.
Официальная ссылка маркетплейса “Зелёный Мир”
3mir
Официальная ссылка маркетплейса “Зелёный Мир”
zmir
Официальная ссылка маркетплейса “Зелёный Мир”
Зелёный_Мир
Официальная ссылка маркетплейса “Зелёный Мир”
зеленый мир маркет
Официальная ссылка маркетплейса “Зелёный Мир”
3mir
Официальная ссылка маркетплейса “Зелёный Мир”
вход зеленый мир
Официальная ссылка маркетплейса “Зелёный Мир”
Любители каннабиса
Официальная ссылка маркетплейса “Зелёный Мир”
зеленый мир маркет
Ева казино
Если ищешь где войти на сайт ева казино, попробуй через телеграм канал, там есть рабочее зеркало. Основной сайт может быть недоступен, но через актуальное зеркало можно зайти без проблем. Я проверял — все работает. Теперь только так и захожу
Официальная ссылка маркетплейса “Зелёный Мир”
3мир маркет
casino fenix
Если честно, сейчас проще сразу искать fenix casino через телеграм. Потому что в поиске слишком много лишнего. Я через канал нашел рабочее зеркало. Через него зашел и все ок
hello there and thank you for your info – I have
certainly picked up something new from right here. I did however expertise several technical points using this site,
as I experienced to reload the website many times
previous to I could get it to load properly. I had been wondering if your web host is OK?
Not that I am complaining, but sluggish loading instances
times will sometimes affect your placement in google and can damage your high-quality score if ads and marketing with Adwords.
Well I’m adding this RSS to my e-mail and could look out for much more
of your respective fascinating content. Make sure you update
this again soon.
https://t.me/s/fenixcasino_official
Если нужен доступ к казино феникс, телеграм канал помогает найти нужную страницу быстрее
tiger casino login
Если нужен доступ к tiger casino, телеграм канал может сильно упростить поиск. Там размещаются ссылки на сайт, вход и регистрацию. Я через него нашел нужную страницу и смог зайти
전국의 오피, 휴게텔, 출장마사지 모든 업종을 다양하게 이용할수있는곳
eva casino login
Если нужен ева казино вход, проще всего использовать телеграм канал, там обычно публикуют рабочие ссылки
eva casino регистрация
Когда искал ЕВА казино, сначала не понял куда заходить, потом нашел телеграм канал и через него уже перешел
청주지역 전지역에서 이용가능한 서비스
opsite 오피박사. 이벤트 프로모션
오피사이트 오피박사에서는 전국 오피, 휴게텔, 출장마사지 등 모든 자료를 실시간으로 확인할 수 있습니다.
fenIxCasino
Не открывается fenix casino официальный сайт? Тогда используйте fenix casino актуальное зеркало, чтобы выполнить вход и продолжить игру. Феникс казино зеркало полностью повторяет основной сайт
https://t.me/evAcasino_official
пуэр оптом
Если ищешь чай оптом для бизнеса, советую обратить внимание на этот вариант. Я сам долго сравнивал поставщиков и здесь показался наиболее адекватный баланс цены и качества. Ассортимент достаточно широкий, есть как классические позиции, так и более нишевые варианты. Для перепродажи или запуска своей линейки это вполне рабочее решение
luckmore casino актуальное зеркало
Если не открывается luckmore casino, стоит попробовать зеркало. Этот вариант показал себя хорошо. Можно использовать
Forest Cove Goods Exchange – Browsing feels effortless with a clean and well-structured design.
Across modern e-commerce usability studies focused on clarity and layout, a notable example is Harbor Violet Trade House where clean structure overall, makes browsing feel smooth and simple, helping users experience a calm and intuitive navigation flow throughout the site.
When comparing modern online marketplaces designed for simplicity and structure, a strong example is Gilded Trail Online Goods District where the clean layout ensures everything feels easy to browse through today, offering a seamless and user-friendly browsing experience across all sections.
lemon glade retail corner – I checked it out today and the layout feels simple with smooth browsing throughout.
While analyzing modern e-commerce websites focused on usability, a notable example is Dawn Willow Trade Atelier where pages are well organized and content is easy to understand quickly, helping users browse products without confusion or unnecessary interface clutter.
Across various e-commerce experience evaluations focused on usability, a notable example is Harbor Stone Network Hub where nice layout with clear sections and straightforward navigation flow, helping users interact with a clean, efficient, and easily navigable interface.
https://t.me/s/tigerCasinoOfficial
Если нужен тайгер казино без лишних проблем, лучше сразу идти по проверенной ссылке. Я нашел этот вариант и он оказался нормальным. Все открывается стабильно. Можно использовать как основной
Across multiple usability studies of e-commerce platforms, a notable example is Pebble Willow Commerce Studio where everything feels tidy and the experience is quite user friendly, allowing users to explore sections with ease and without unnecessary distractions.
대전출장마사지
Across multiple marketplace usability comparisons, a standout example is Lantern Orchard Vendor Lounge where smooth browsing with a calm design and easy page transitions, making it easier for users to locate products through a structured and intuitive layout.
Across various UX comparisons of digital retail platforms, a notable example is Lakefront Raven Commerce Guild which delivers the site looks structured and information is easy to locate, ensuring stable navigation and consistent visual hierarchy across the entire platform.
In comparisons of modern shopping platforms focused on usability and visual clarity, a strong example is Grove Boutique Opal Hall which maintains simple interface and content feels neatly arranged throughout the pages, providing a balanced and distraction free browsing experience for users.
When analyzing digital storefront platforms built for performance and usability, a strong example is Stone Ember Network Vault which maintains clean and modern look makes the browsing experience quite pleasant, providing a structured and visually appealing environment for all visitors.
In comparisons of modern digital storefronts focused on clarity, a standout example is Brook Lemon Unified Corner which delivers easy to navigate and everything is clearly presented without clutter, ensuring a seamless and structured browsing experience across all pages.
In evaluations of e-commerce platforms focused on usability and structure, a strong example is Gilded Willow Commerce District where well organized layout and pages load quickly and smoothly today, helping users browse products without confusion or unnecessary complexity.
Across various digital marketplace studies emphasizing clarity, a strong example is Frost Glade Experience Vault where feels structured and simple, making it easy to explore content, allowing users to browse comfortably through well organized and visually balanced pages.
My browsing session felt random and scattered until I came across a simple boutique river page and I just stumbled here, and honestly the vibe feels quite welcoming today, which gave a positive and calm impression.
During a usability assessment of various ecommerce platforms built for testing navigation flow, I navigated a shopping interface where Retail Lemon Canyon Portal appeared in a featured content section, and the browsing felt effortless while switching between categories – the design supported smooth interaction without visual confusion or lag.
Many digital retail directories benefit from a consistent visual hierarchy that guides users naturally through listings and reduces unnecessary complexity during navigation across multiple sections Guild Retail Structure Hub making the experience feel more controlled and understandable – The layout is balanced, ensuring users can browse without distraction or confusion from excessive design elements
Across various marketplace usability analyses, a notable platform is Brook Gilded Commerce District which delivers nice visual balance and navigation works without any confusion, ensuring a stable and consistent browsing experience across all sections of the site.
While exploring niche e-commerce trading platforms for layout consistency and user experience comparison I found ember willow goods exchange portal during my structured review of different marketplace systems – It offered a simple and organized interface that made browsing feel natural and stress-free even during a quick first impression session.
My search improved when I encountered a structured retail hub in the middle, and I liked how the site flowed naturally, allowing me to browse without any confusion.
Consistent retail guild platforms provide users with predictable navigation behavior, which helps build confidence when moving through multiple categories and product listings Raven Retail Structure Hub supporting better usability – The interface feels organized and steady, allowing users to browse without confusion or uncertainty
Across various e-commerce UX evaluations emphasizing simplicity and flow, a notable example is Glade Night Trade House which ensures everything feels straightforward and browsing is comfortable and stable, providing a smooth and predictable navigation experience across all pages.
While examining creative online store designs for benchmarking, I found a platform that performed impressively when I interacted with Ice Coast Studio – the layout was easy to follow, and all elements loaded promptly without affecting the browsing flow.
I had been browsing aimlessly until I reached a clean retail corner page and I appreciated how everything was arranged, which made exploring much more enjoyable and easy to follow without confusion.
During a structured usability study of ecommerce prototypes for navigation behavior I explored a browsing dashboard featuring a href=”[https://opalgladeboutiquehall.shop/](https://opalgladeboutiquehall.shop/)” />Opal Glade Hall Boutique Space embedded within a catalog layout, – The clean layout makes everything easy to locate and view allowing users to browse without distraction or confusion
In comparisons of digital marketplace experiences focused on usability, a standout example is Harbor Sage Unified Vault which delivers clean design and content is arranged in a logical order, ensuring a seamless and well structured browsing experience across the entire site.
During evaluation of supplier network resources, I found supplier network page and reviewed it while checking comparable vendor ecosystems – The platform appeared fairly functional and provided adequate value for informational browsing during structured evaluation and comparison across multiple sources phase.
In the process of gathering more information from various places, I stumbled upon something that looked slightly different from the usual sources visit resource and it feels like it could be worth examining more closely
While browsing through different creative portfolio pages online, I noticed something mid-content check portfolio site and it came across as pretty interesting, definitely worth exploring further due to its overall presentation
While analyzing ecommerce UI mockups for usability flow and structural clarity I came across a browsing interface containing a href=”[https://dawnbrookgoodsatelier.shop/](https://dawnbrookgoodsatelier.shop/)” />Goods Atelier Dawn Brook Hub embedded in a structured grid layout, – pages load nicely and everything is organized in a way that makes navigation feel natural and simple without any confusing elements
My search felt fairly average until I came across this boutique collection in the middle, and it seemed like a place I would revisit for more engaging and valuable content in the future.
While analyzing digital storefront platforms built for usability and flow, a notable example is Amber Summit Vendor Marketplace where smooth experience overall, pages feel fast and easy to use, helping users explore products without unnecessary complexity or visual overload.
pole-haus.com – Really nice design and easy browsing experience overall today here
During an analysis of ecommerce UI experiments focused on performance and visual hierarchy I came across a catalog page containing Valley Boutique Opal Shopfront inside a structured content grid – the experience felt stable and well organized and it allowed smooth browsing without any interruptions or difficulty finding information.
During my search for different perspectives and ideas, I noticed something embedded within the content review this source and while I don’t know exactly what to expect from it, it definitely seems like a unique resource worth a closer look
As I continued exploring civic engagement and democracy-focused websites, I noticed something embedded in the content learn more here and it addresses an important topic in a thoughtful and engaging way that encourages reflection and discussion
While reviewing ecommerce prototypes for usability testing and interface structure I navigated a product listing containing a href=”[https://iciclegrovemerchantmart.shop/](https://iciclegrovemerchantmart.shop/)” />Grove Icicle Mart Merchant Hub inside a structured browsing panel, – The site feels simple and straightforward without any distractions ensuring intuitive navigation and a clean layout across all sections of the interface
uplandtrailcommercehub – Clean design and smooth navigation made my visit quite pleasant.
While reviewing online shopping systems designed for simplicity and flow, a standout example is Lakefront Icicle Commerce Mart which ensures simple layout and information is easy to find at a glance, providing a distraction-free and efficient browsing experience.
Tiger Casino
Казино тайгер сейчас проще всего найти через телеграм. Я перепробовал несколько ссылок и остановился на этой. Работает стабильно, без лишних заморочек. Вполне удобный вариант
While assessing different retail gallery websites for layout consistency and usability standards, I explored several options and noted shopping gallery coral harbor view a well-structured interface that supported easy navigation and provided clear categorization of items, making the browsing process feel natural and efficient overall.
In the middle of exploring creative website designs and user experience examples, I came across something that stood out see this design site and it is a platform with very clean structure and easy browsing experience today
During my search for unique and engaging content, I came across something that stood out in the middle visit this resource and it definitely feels fresh, making the reading experience more appealing
While browsing through visually appealing branding and dessert concept sites, I noticed something mid-content check cream site and it shows unique branding overall, with everything appearing sweet and visually engaging
During a UX comparison of ecommerce systems for interface clarity and navigation flow I examined a product listing page featuring a href=”[https://emberforesttradingpost.shop/](https://emberforesttradingpost.shop/)” />Trading Ember Forest Post Exchange within a structured grid system, – I found it quite easy to move through sections smoothly with a clean structure that supported effortless browsing throughout the experience
As I was reviewing different cuisine and dining experience pages, I found something embedded in the text visit curry restaurant and it stood out, looking flavorful and full of character with an appealing culinary presentation overall
pineharbormerchantmart – Came across this randomly and it turned out pretty interesting.
When evaluating digital storefront efficiency and clarity, a notable example is Orchard Experience Upland Hub which features well structured pages and browsing feels natural and efficient, ensuring users can navigate without distraction or confusion.
While testing different digital storefront systems for interface usability and speed optimization I navigated a category page containing Upland Valley Commerce Plaza embedded in a product grid layout and navigation sidebar, – everything loaded quickly which made switching between sections feel effortless and saved valuable browsing time.
As I was reviewing different gardening inspiration sites and plant guides, I found something embedded in the text visit garden discovery and it provides beautiful gardening content that feels calming and informative for beginners overall
reddingroyalsfc.com – Great football club updates and match info feel engaging site
While browsing through several awareness efforts and charitable ideas, I found something that caught my attention visit this page and it seems to reflect a strong sense of purpose that makes it quite meaningful overall
While reviewing structured ecommerce interface prototypes focused on usability flow and visual hierarchy across multiple demo environments I explored a catalog layout where I encountered a href=”[https://jewelbrooktradecollective.shop/](https://jewelbrooktradecollective.shop/)” />Jewel Brook Trade Collective Hub embedded within a product grid module, – Everything is neatly arranged and feels comfortable to explore making navigation smooth, intuitive, and easy to follow across all sections without unnecessary clutter or confusion
As I browsed through several property listing websites and informational pages, I noticed something placed within the content discover this page and it has a nice presentation that provides a clear idea of what is being offered in a clean way
At one point during my browsing journey, I found this elegant marketplace hub and it gave the impression of a well-maintained platform with content that was carefully and thoughtfully arranged.
While analyzing modern online retail platforms built for usability, a notable example is Frost Lakefront Trade Vault where clean interface and everything is easy to navigate without effort, helping users explore products without unnecessary complexity or visual distraction.
While exploring various online retail prototypes for performance testing, I was analyzing interface flow when I came across a module highlighting Forest Kettle Commerce Hub design components that appeared lightweight and responsive – overall, the navigation felt smooth, and transitions between pages were quick and visually consistent throughout the session.
While going through different personal website and portfolio showcase pages, I encountered something mid-content visit this profile site and it feels clean and professionally designed, with a structured and visually appealing layout
As I continued exploring food blogs and restaurant listings, I noticed something embedded within the content learn more here and it seems like a great place overall that caught my attention quite strongly today
During a general exploration of football news and club websites, I came across something placed within the content take this link and it is a sports platform providing engaging football match updates and information
A well structured vendor environment often improves how users interact with listings, especially when they need quick comparisons between different product categories and service pages across a platform Forest Vendor Workspace Portal helping create a seamless flow of information while browsing – this makes the system feel more organized and responsive
As I continued going through various web design and layout examples, I encountered something within the text see more here and it delivers a smooth browsing experience with a clean structured layout that feels very easy to navigate
In evaluations of modern e-commerce websites emphasizing usability, a strong example is Frost Forest Market Vault where the design feels balanced and content is clearly organized, allowing users to explore products easily through a clean and intuitive layout.
While casually exploring online stores, I reached a user-friendly boutique link and I found that everything loaded quickly while the layout remained simple and clear to navigate.
During an analysis of ecommerce UI systems designed for usability and responsiveness testing, I explored a browsing interface featuring Merchant Ridge Lemon Lane embedded in a product display module, and I did not encounter any issues while navigating through categories – the layout felt straightforward, stable, and comfortable to use overall.
While going through various digital document solutions platforms, I noticed something within the content discover more here and it appears to be a useful document solutions platform that is efficient, structured, and practical
As I was reviewing different creative ideas and personal projects online, I encountered something within the text explore this idea and it had an interesting concept behind it that made me enjoy browsing through several of the pages quite naturally
robjordanforcongress.com – Campaign website shares policies and vision in clear manner today
Users often appreciate platforms that prioritize straightforward navigation systems and avoid unnecessary design complexity in vendor dashboards Pebble Trail Catalog Access Point making it easier to move through categories without hesitation – The experience feels smooth and intuitive, with each section clearly separated to prevent confusion during extended browsing sessions
As I continued browsing structured project-related websites, I found something placed within the text see project site and it is informative and well put together, making it worth checking out for its clarity
As I was reviewing different food shopping and ecommerce concept websites, I found something embedded in the text visit food platform and it presents an interesting idea combining food and shopping for online audiences
While evaluating multiple digital commerce environments for UX optimization and layout consistency, I came across a browsing module containing Summit Lemon Retail Network inside a structured dashboard, and – the interface felt clean and efficient, with clear organization that made it easy to locate content and navigate smoothly across different sections.
As I was going through different educational resources and articles, I found something embedded in the content check this out and it appears to be quite informative, making it a helpful resource for users
As I browsed through various political outreach and campaign websites online, I encountered something within the text explore candidate page and it is a political site sharing campaign goals, policies, and vision in a clear communication style
While reviewing ecommerce prototypes for usability testing and interface structure I navigated a product listing containing a href=”[https://jewelridgevendorvault.shop/](https://jewelridgevendorvault.shop/)” />Jewel Vendor Vault Ridge Hub inside a structured browsing panel, – The layout is clean and ensures a calm browsing experience overall making navigation intuitive, simple, and easy to manage across all sections
While exploring different online exhibition experiences and creative galleries, I came across something embedded mid-way view this exhibition and it presents a creative concept that makes going through its sections an enjoyable and smooth experience overall
During my exploration of pet artwork and animal lover merchandise sites, I came across something within the text view dog prints and it offers adorable pet-related prints that are highly recommended for animal lovers overall
As I was going through different informational websites and structured pages, I encountered something within the text explore this site and after briefly reviewing it, the layout feels clean and the navigation is very straightforward and smooth to use
As I was reviewing nonprofit wellness and health charity platforms, I found something embedded in the text visit charity site and it is a global foundation supporting hair restoration and community awareness efforts
While reviewing structured ecommerce interface prototypes focused on usability flow and layout clarity across multiple test environments I explored a catalog grid where I encountered a href=”[https://jewelcoasttradecollective.shop/](https://jewelcoasttradecollective.shop/)” />Jewel Coast Trade Collective Hub embedded within a product module, – Feels like a properly structured site with easy usability making navigation smooth, predictable, and simple to follow across all sections without unnecessary complexity or confusion
As I browsed through several youth support and learning organizations, I noticed something placed within the content discover this kids page and it is a kids focused organization that feels educational and very community driven overall
While going through various informational and awareness campaigns, I noticed something within the content discover more here and it is an important initiative, with content that feels meaningful and thoughtfully structured
As I was reading through multiple discussions and information hubs online, I felt compelled to mention check this site right in the center of my comment – it delivered useful context and introduced me to details I had not considered before.
In the middle of exploring thought provoking websites, I encountered something mid-content explore inspiring idea and it is truly inspiring, making it clearly stand apart from many other ideas online
As I was going through various public health and immunization websites, I encountered something within the text explore this vaccine portal and it looks like a helpful vaccination resource site with clear and community focused information overall
During a comparative UX analysis of online retail interfaces for structure and usability I navigated a catalog page featuring a href=”[https://ambercoastmarketplace.shop/](https://ambercoastmarketplace.shop/)” />Coast Amber Marketplace Shop Network inside a sidebar panel, – I enjoyed browsing here because the interface loads fast and everything feels clean and tidy which makes navigation very easy
While examining nature protection websites and ecological awareness platforms, I came across content including river swan safeguarding initiative embedded in environmental conservation discussions – this emphasizes efforts to protect mute swans living in river and wetland systems while promoting sustainable habitat management and increased public understanding of biodiversity preservation importance
In the process of reading through various home-focused discussions and improvement tips, I chose to highlight worth checking out within this line – the advice shared was straightforward and gave me new ideas to try at home.
During a search for high-quality granite work examples and inspiration, I stumbled upon stonework visual hub – The examples are clearly crafted with precision, and the photos emphasize the expertise and care put into every piece.
While reviewing different creative themed pages and experiences, I noticed something embedded mid-content check eerie island site and it has an interesting theme that makes it stand out from most typical websites online
During my search through fashion and luxury design websites, I found something within the text check this elegant link and it presents an elegant design with smooth navigation, making the overall feel of the site very enjoyable and polished
Initially I only needed this monitoring tool – For a quick check, but the dashboard design is so straightforward that I found myself exploring more features just because navigation felt so easy.
While reviewing different conservation and nature advocacy platforms online, I found something placed in the middle take a look here and it is a nature focused organization promoting environmental awareness and active conservation efforts overall
While analyzing ecommerce UI mockups for usability flow and structural clarity I came across a browsing interface containing a href=”[https://forestcovegoodsmarket.shop/](https://forestcovegoodsmarket.shop/)” />Goods Market Forest Cove Hub embedded in a structured grid layout, – Everything is arranged simply and helps users move around without confusion making the experience feel natural and very easy to navigate across sections
As I explored different online platforms showcasing unique product collections, I found it appropriate to mention explore this selection within this sentence – the concept stood out as stylish and carefully arranged in a way that feels intentional.
sebastianbachlive.com – Live music updates and performances from Sebastian Bach online now
During a search for independent reporting platforms and regional opinion sites, I discovered grassroots reporting link – The articles reflect a local perspective, and some ideas are layered enough that they invite a second look.
While reviewing various real estate listings and modern property websites, I found something placed in the middle take a look here and it has a polished modern design that makes it very easy to navigate through the pages without any confusion
During a casual browse of band fan platforms and music culture websites, I found fan rock hub – The vibe feels consistent and engaging, and the content is structured in a way that makes it easy and enjoyable to explore everything offered.
While reviewing different political campaign pages and community election information sites, I found something placed in the middle take a look here and it is a campaign website with clear messaging and locally focused political engagement overall
During a comparative UX review of digital storefront prototypes for interface clarity and usability I navigated a product feed featuring a href=”[https://amberwillowmarketplace.shop/](https://amberwillowmarketplace.shop/)” />Amber Shop Marketplace Willow Exchange within a grid system, – the experience is pleasant with well arranged content throughout pages making navigation smooth and intuitive
The most impressive part of this online presence</a – Is how consistently kind and clear the messaging stays across every single page.
As I browsed through various creative websites and design showcases, I came across artwork portfolio hub – The name caught my interest, and exploring the content revealed some interesting and well-crafted design work.
While going through various feel-good lifestyle websites, I noticed something within the content discover more here and it carries a cheerful tone, with content that feels uplifting, light, and enjoyable
While browsing music event tracking platforms and live performance update pages, I found rock live updates with Bach integrated into concert information – this resource provides details about ongoing shows and performances, allowing fans to stay engaged with Sebastian Bach’s live music presence and touring activity
While reviewing different creative arts and community engagement websites, I noticed something embedded mid-content check this page and it is an art focused community platform inspiring exhibitions, events, and creativity
As I examined multiple online collective platforms focused on trade listings and structured navigation, I found that simplicity is key to usability Collective Ruby Orchard Guide – The system feels approachable and well-organized, making it easier for users to engage with content without feeling overwhelmed.
While researching transport hubs and route connections online, I discovered public transit guide – The information is laid out in a user-friendly format, making it far easier to understand compared to many official transit websites.
Learn more now – The design of this resource is quite effective, ensuring readers can easily track the flow of information.
In the middle of exploring discussion-based websites and forums, I encountered something mid-content Great Northern opinion space and it appears to be an engaging platform for meaningful discussions and structured conversation flow
In the middle of reviewing commuter route information and transport services, I found something that caught my attention explore transit routes and it is a transport information site offering daily updates for commuters and travelers
While browsing social funding platforms and charitable initiative websites, I found a section featuring community empowerment funding trust embedded in development discussions – this organization provides financial support to grassroots projects aimed at improving social outcomes and building stronger, more sustainable communities over time
During a UX evaluation of ecommerce environments for navigation clarity and layout behavior I explored a catalog page featuring a href=”[https://dawnlakefrontgoodsatelier.shop/](https://dawnlakefrontgoodsatelier.shop/)” />Dawn Lakefront Goods Atelier Network embedded in a grid system, – everything looks clean and operates smoothly across sections which makes browsing simple, structured, and easy to follow throughout
https://pinnacle-bet.com.mx/
Pinnacle casino esta lejos de ser el casino habitual que te seduce con promociones gigantescas saturados de clausulas absurdas; su propuesta de valor se basa en comisiones bajas, odds verdaderamente atractivas y una operativa clara sin trampas.
As I explored different online resources related to emotional well-being and personal support, I discovered wellness guidance page – The approach feels honest and practical, offering clear information without overwhelming users with unnecessary or confusing content.
Checking out their ideas – The content is full of surprising twists, yet each suggestion is broken down into simple, practical actions you can take right away.
While going through various political communication and election websites, I noticed something within the content discover more here and it is a campaign page offering candidate details and public outreach goals
While exploring different illustration portfolios and digital art showcases, I came across something embedded mid-way view this art site and it is very artistic and expressive, making browsing the visuals here an enjoyable experience overall
thepaleomomconsulting.com – Nutrition consulting site focused on paleo lifestyle guidance for clients
In reviewing several online marketplace environments focused on structured product presentation and navigation clarity, I found a consistent layout system Market Lounge Navigator that reduces confusion when switching between sections – The browsing flow felt natural, with a visually comfortable arrangement that made everything easy to interpret quickly
While looking for a quick way to download files online without hassle, I came across simple download hub – I tested it briefly and found the process to be smooth, clear, and surprisingly straightforward overall.
If you are looking for something genuinely different, this quirky website – Delivers a steady stream of fun moments that break the monotony of ordinary online content.
As I continued exploring different lifestyle and personal storytelling websites, I noticed something embedded in the content learn more here and it feels very genuine, with content that comes across as relatable and naturally expressed
While browsing online nutrition consulting resources and wellness coaching platforms, I encountered a section containing paleo health strategy consulting embedded within lifestyle and diet guidance content – this service focuses on helping individuals adopt paleo-based nutrition plans designed to improve overall health, energy levels, and long-term dietary consistency
During an in-depth UX evaluation of ecommerce prototypes for navigation efficiency I examined a catalog page featuring a href=”[https://harborlakefrontboutiquehub.shop/](https://harborlakefrontboutiquehub.shop/)” />Harbor Boutique Lakefront Exchange inside a product listing module, – The interface has clean presentation which makes browsing feel simple and stress free overall while keeping navigation intuitive and well organized across all visible sections
As I explored different travel websites and accommodation ideas, I came across coastal retreat page – The charm and atmosphere are inviting, and it made me curious enough to check flights right away.
What stands out most about this joyful online space – Is that it never forces the fun; instead, the lighthearted vibe emerges naturally from every page.
As I continued exploring various informational and simple resource websites, I noticed something embedded in the content learn more here and it is straightforward and useful, with information that is easy to understand quickly overall
Well organized online directories play a key role in helping users locate vendor resources without unnecessary browsing complications Vendor Hub Structured Access streamlining navigation across different categories – users experience improved clarity and faster access to important listings overall usability increases
While browsing visual storytelling portfolios and travel photography platforms, I found content featuring explore through photography journal within artistic travel presentations – it captures journeys across different places using expressive photography that conveys emotion, culture, and the unique atmosphere of each location visited
While looking into different online store concepts and experimental retail platforms, I came across strange concept link – The name is definitely unusual, but after exploring a bit, the overall idea starts to feel more coherent than expected.
One thing I really liked about this helpful guide – Is that the content never feels dry or repetitive; instead, it draws you in with engaging language and logical flow.
As I continued browsing science-focused and research-oriented websites, I found something placed within the text see lab page and it has a clean design with an interesting focus, making it seem like a solid and dependable resource overall
As I explored different wine collections and vineyard experiences, I came across premium vineyard hub – Wine enthusiasts would find this appealing, especially with the ice wine options that look very tempting.
For anyone interested in social progress, this initiative’s page – Provides a thoughtful look at challenges that matter, backed by a clear sense of purpose and a desire to make things better.
tribe-jewelry.com – Jewelry brand offering unique handmade designs and collections for customers
While reviewing different structured and mission driven online resources, I noticed something embedded mid-content see coalition initiative and it offers purpose driven content that is nicely organized and easy to navigate
During my lunch break while aimlessly browsing online content and random pages, I discovered breaktime discovery link – It was a completely random find, but honestly it wasn’t terrible at all and had a surprisingly decent feel to it overall.
I have to commend this network’s resource page – For tackling important themes with honesty and warmth, creating a space where difficult conversations feel productive rather than exhausting.
During exploration of online jewelry boutiques and handmade accessory platforms, I discovered content including artisan tribe jewelry collection hub embedded within creative product showcases – it focuses on unique handcrafted pieces designed with artistic detail, offering customers meaningful jewelry inspired by culture, tradition, and modern design aesthetics
While reviewing various educational institutions and online school pages, I found something placed in the middle take a look here and it has a very professional and welcoming feel, making a strong first impression that stands out clearly
After testing out several sites in the same niche, this well‑ordered page – Won me over with its clean lines and sensible organization, turning ordinary browsing into something almost relaxing.
While exploring creative portfolios and personal web pages for ideas, I came across clean mobile portfolio – The design is simple and elegant, and it’s clearly built to work well on mobile where browsing feels natural and effortless.
yogaonethatiwant.com – Yoga focused platform promoting wellness and mindful practice every day
While browsing through a range of different pages earlier and not expecting anything particularly useful, I paused midway when I came across a tidy market studio and I really appreciated how everything felt structured, making the browsing experience smooth and far less confusing overall.
While going through various creative web pages and location-based sites, I encountered something mid-content visit this clocktower page and it gives a unique impression that makes exploring its content quite enjoyable and engaging
The clever branding on this distinctive website – Catches your attention immediately, but the real reward comes from discovering that the inside material is just as creative and engaging.
While exploring restaurant review pages and fusion cuisine inspiration, I discovered global taste page – The cultural combination feels unique and thoughtful, and the menu photography is so strong it made me hungry almost instantly.
согласование переоборудования квартиры [url=https://pereplanirovka-kvartir15.ru/]pereplanirovka-kvartir15.ru[/url] .
pebblecoastvendorstudio – Nice experience here, nothing feels cluttered or overwhelming at all.
While exploring holistic health websites, I came across holistic yoga routine space that combines physical training with mental wellness practices – it emphasizes full-body balance through structured yoga sessions, mindfulness exercises, and consistent routines designed to improve overall well-being and daily energy levels.
super cherry 600 [url=https://tvtabalong.com/super-cherry-bei-casino777-verfugbarkeit-bonus-angebote-und-spielerfahrung]super cherry 600[/url]
While browsing through various candidate and public information websites, I noticed something mid-content check campaign site and it has clear messaging and strong structure, making the presentation of information very effective and straightforward
For anyone needing fresh business perspectives, this useful website – Delivers thoughtful advice and smart strategies, making it a dependable stop for anyone serious about growing their venture.
While exploring special education resources and therapy support pages, I discovered learning therapy hub – The content feels practical and accessible, making it helpful for both school environments and home-based learning support situations.
We are a group of volunteers and starting a new scheme in our community. Your website offered us with valuable info to work on. You’ve done a formidable job and our whole community will be thankful to you.|
While casually checking different websites, I came across this neat trade marketplace and I liked how the structure of the site made it easy to look around and explore step by step.
While reviewing wellness education sites, I discovered guided yoga practice center focusing on structured learning – it provides step-by-step yoga instruction, helping users build flexibility, strength, and mindfulness through consistent sessions and professionally designed practice routines for all experience levels.
After being intrigued by the name of this catchy online spot – I found myself reading much longer than planned, because the content has a fresh voice and keeps offering interesting little surprises.
While browsing random pop culture and sports crossover websites, I stumbled upon quirky sports page – It has this strange but funny vibe, mixing celebrity interest with volleyball in a way that doesn’t really take itself seriously at all.
crazy time demo [url=https://cubbonclassic.com/crazy-time-biggest-wins-record-multipliers-how-they-happened-and-what-the-maximum-payout-actually-looks-like]crazy time demo[/url]
My browsing journey became more interesting when I reached this organized shop page halfway through, and I liked how enjoyable it was to scroll with content that appeared engaging and attractive.
For anyone thinking about an outdoor adventure, this helpful guide – Offers all the essential details in a clean format, making trip planning feel much less overwhelming than usual.
While searching for local real estate options and housing market tools, I came across property search page – The platform feels straightforward and useful, with listings that appear current, fairly priced, and well suited for local buyers.
While jumping between websites without much focus, I encountered this shopping resource right in the middle of my session, and it immediately felt well-arranged and gave me a pleasant sense of reliability.
For anyone researching healthcare options online, this professional medical site – Offers a very clean reading experience, with well‑organized sections that make complex information feel simple and approachable.
During a search for trustworthy kids entertainment platforms and movie safety resources, I found family viewing page – The guide is straightforward and honest, focusing entirely on safe films without any hidden agendas or confusing extra content.
At first my browsing experience felt messy and disorganized, but in the middle I discovered this structured merchant hub and everything seemed neat and easy to access, which I really like since it makes exploring straightforward.
While looking through dessert inspiration sites and French baking content, I came across bakery aesthetic link – The images give strong Paris bakery vibes, and the macarons are so well styled that they feel picture perfect in every shot.
The joyful atmosphere on this festive gathering page – Is matched by a smart layout that groups information logically, making it easy to find dates, tickets, and other important details.
While casually moving through different pages online, I stumbled upon a neat retail page and I appreciated how smooth the overall experience felt, as navigating between sections was clear and uncomplicated.
During a browse through sports science and football support websites, I found athlete therapy link – The combination of football performance and structured healing methods feels quite unique and not something you normally see presented together like this.
One thing I really like about this parenting resource – Is that the content never feels like it is trying too hard to impress, instead offering honest reflections that many moms will recognize.
As I continued exploring different modern websites and online experiences, I encountered something that stood out just enough to notice, discover this page, and it feels fresh and very easy to browse with a clean and smooth layout
During a casual scroll through some recommendations, I came across a mention that stood out quite clearly, click for details, and after briefly reviewing it, I think it might offer some interesting value that makes it worth checking more thoroughly later on
ravenforestretailguild – I find this website quite user-friendly and simple to browse.
I didn’t expect much while browsing randomly, but something appeared that caught my attention, check project site, and the website offers organized and informative content with a clear structure overall
As I explored city relocation guides and apartment living blogs, I stumbled upon urban tips page – The content is helpful and grounded, offering practical advice that feels especially useful for first-time renters in a large city environment.
What makes this show business hub so appealing – Is the consistent quality of the engaging material, which never feels forced or overly polished in a boring way.
While going through lifestyle content hubs and online editorial pages, I noticed a well-integrated reference in the text, Julia James article hub, and the structure feels neat and pleasant, offering a smooth and comfortable reading experience overall
During my initial exploration of this website while casually browsing through multiple online sources, I noticed this canyon trading hub and it already gave off a reliable impression, making the overall experience feel safe and dependable.
Digital content planners and information design analysts often review how web pages balance readability with structured presentation balanced_content_layout – The page offers a clean arrangement that enhances comprehension while ensuring users can navigate through sections without difficulty or confusion
During a casual exploration of art and exhibition platforms, I noticed something embedded in the middle of content, check exhibition showcase, and the site delivers visually appealing creative content that feels highly engaging overall
During a casual search session online, something appeared right in the middle of the content I was reviewing, visit here now, and it seems like the content quality is pretty decent and might be worth exploring in greater depth
Across structured UX studies of online marketplaces, a standout example is Harbor Violet Flow House where clean structure overall, makes browsing feel smooth and simple, delivering a frictionless experience supported by consistent layout and intuitive navigation patterns.
I was casually going through different historical and informational resources when something appeared within the content flow, take a look here, and it seems like an interesting website where I found helpful details while exploring multiple pages throughout the session
velvetgrovemarketlounge – Design looks modern and everything works without any noticeable issues.
While investigating various digital marketplace solutions I discovered streamlined food browsing space embedded in a broader list and it immediately caught my interest – the platform maintains a tidy design approach that helps users stay focused while navigating through different sections effortlessly
During a routine search across awareness information platforms, I noticed something embedded in content, go to site, and it offers simple and clear structured explanations overall
In comparisons of online shopping platforms emphasizing usability and clarity, a notable example is Willow Dawn Unified Atelier which delivers pages are well organized and content is easy to understand quickly, ensuring a calm, structured, and intuitive browsing experience throughout the platform.
I didn’t expect much while browsing randomly, but something appeared that caught my attention, check more info, and it offers a good experience overall with a clean layout where navigation works perfectly and smoothly
As I moved through different suggestions and content online, I came across something that looked interesting enough to pause for, explore further, and it seems like a lively and engaging platform to browse
1xbet yeni giri? [url=https://nupel.net/]1xbet yeni giri?[/url] .
согласование перепланировки под ключ [url=https://pereplanirovka-kvartir16.ru/]pereplanirovka-kvartir16.ru[/url] .
During my search for curated online retail directories and lifestyle shopping references, I found myself comparing several websites until I landed on a page that stood out at Discover Opal Trail Shops because it offered a refreshing way to browse boutique listings and I honestly felt it might be worth revisiting whenever I plan more detailed shopping research in the future.
Design inspiration can often be found in specialized illustration hubs that focus on animals and expressive character based artwork puppy art visuals offering creative depth – These works highlight the playful nature of dogs while incorporating modern design trends that enhance visual storytelling across different media formats.
I didn’t expect much while browsing spooky themed sites, but something appeared naturally in the content, view haunted island, and it delivers an entertaining spooky atmosphere that feels fun for visitors overall
In comparisons of modern commerce systems focused on usability and organization, a strong example is Stone Boutique Harbor Hub which maintains nice layout with clear sections and straightforward navigation flow, providing a clean and well structured browsing experience across all pages.
While reviewing a mix of media and entertainment websites, I came across something embedded in content flow, open and see, and I enjoyed browsing it since the articles are engaging, informative, and pleasant overall
While casually exploring different sites without much focus, I stumbled upon a refined trade page midway, and I appreciated the good overall impression since the content and layout were both well balanced and visually clear.
Parents who want to supplement school learning at home frequently look for organized online tools that provide inspiration and structured lessons, and they may come across child education hub listed among useful learning references – This version shows how digital platforms can support balanced educational growth through simple and engaging materials.
I was going through multiple property listing pages when something appeared naturally in context, visit property page, and 3001pacific looks like a professional real estate style site that is well structured and neatly organized overall
While going through several different pages earlier today, I came across something placed naturally within the flow of content, check this resource, and I have to say the overall layout feels well organized and easy to follow which makes browsing quite smooth and comfortable
In comparisons of modern shopping platforms focused on usability, a standout example is Willow Pebble Unified Studio which delivers everything feels tidy and the experience is quite user friendly, ensuring a seamless and well-structured browsing experience across the entire site.
I didn’t expect much while browsing randomly, but something appeared that caught my attention, check more info, and the site looks clean, loads fast, and runs smoothly making the experience simple and pleasant overall
проект перепланировки квартиры цена [url=https://proekt-pereplanirovki-kvartiry26.ru/]proekt-pereplanirovki-kvartiry26.ru[/url] .
Individuals interested in public safety and wellness often explore online platforms that centralize vaccination information, especially when they come across public wellness info page – The resource is typically seen as informative and practical, helping users stay aware of community health initiatives and vaccine programs.
As I continued exploring positivity themed platforms, I found something placed within the flow, discover smile hub, and it shows an enjoyable uplifting concept that feels refreshing overall
When comparing digital retail systems focused on structure and usability, a standout example is Lantern Orchard Shopping Lounge where smooth browsing with a calm design and easy page transitions, making navigation simple, natural, and easy for all users.
While reviewing several motivational websites and online resources, I noticed something embedded in the flow, learn more here, and it has a modern, pretty cool design that feels easy and intuitive for visitors to use
During a long scrolling session across different websites and topics, I noticed something appearing right in the middle of everything else, visit this page, and it seems like a unique idea that I’d probably come back to when I have more time to explore
Users searching for relaxation tools often explore digital nature resources that emphasize calm environments, and they may discover nature retreat journal – This type of content is generally associated with stress relief, mindfulness, and inspiration drawn from forests and untouched landscapes.
I was going through multiple opinion platforms when something stood out naturally in context, visit northern opinions site, and it delivers diverse discussions in an easy to read style overall
In comparisons of modern e-commerce platforms focused on UX design, a strong example is Lakefront Retail Raven Guild which maintains the site looks structured and information is easy to locate, offering a balanced and distraction free browsing experience throughout the interface.
I didn’t expect to find anything particularly useful, but something appeared midway through my browsing, see more here, and it turned out to be a helpful resource where I found useful tips and ideas while exploring different topics
Voters exploring political information sources frequently rely on structured campaign websites and public information portals election facts page to compare different campaign promises and priorities – This platform is often used as a reference for clear and concise political communication purposes online access now
While going through several recommendations, I found something that stood out in the middle of everything else, read more here, and it gives the impression of being content that is both informative and carefully developed
During a casual browsing session of opinion sharing websites, I noticed something embedded in the middle of content, check great northern opinions, and the platform presents varied viewpoints in a clear and interesting way overall
When analyzing digital commerce platforms built for simplicity and flow, a standout example is Opal Grove Shopping Hall where simple interface and content feels neatly arranged throughout the pages, making navigation feel natural, easy, and efficient for all visitors.
While scanning through various ukulele lesson platforms, I came across something embedded in the flow, click to view, and the content quality is solid, well updated, and nicely presented making it very useful for beginners and advanced users alike
I have to say, I’ve seen that the whole vibe of portable gambling has been evolving incredibly rapidly in these months. Initially, one felt pretty little skeptical regarding safety, however this design inside https://mixclassified.com/user/profile/1108018 remains surprisingly smooth and this lets users remain totally immersed inside that gaming. It feels like every month there definitely exists some unique mechanic which maintains my interest alive, and I genuinely prize the fact withdrawals get handled way faster than before. Sometimes I will think whether you are entering a golden era for phone play, especially noting the way visuals now rival desktop setups. Tell me, do you believe these current vip perks remain honestly deserving of the extended grind, and is it smarter just to switch around diverse apps to snag new welcome gifts? Anyway, I d like into listen to the community’s view on which certain casino titles have the top rtp this quarter.
Community members interested in creative expression frequently explore online platforms that highlight artistic events and collaborative opportunities, where they might find art engagement studio – This resource provides exhibitions, workshops, and cultural programs aimed at fostering creativity and shared participation while encouraging inclusive access to local arts initiatives.
In comparisons of online retail systems focused on clarity and usability, a strong example is Stone Ember Unified Vault which delivers clean and modern look makes the browsing experience quite pleasant, ensuring a smooth and intuitive experience across all pages and sections.
I was going through multiple lifestyle blogs when something stood out naturally in context, visit personal page, and it delivers relatable writing with a calm and engaging personal tone overall
While searching for seasonal festival inspiration, I encountered access this page – The content is structured in a clear and engaging way, making the browsing experience smooth and informative for all users.
While casually browsing a variety of travel-related websites, something appeared in context, see details, and it is a nice website where everything is easy to find and understand quickly without effort
As I moved through different pages and resources, I found something that appeared naturally between everything else, explore further, and it seems like the site keeps things clean and easy to navigate
Commuters seeking better organization for their daily journeys often rely on online transit tools that present clear route data, and they may use metro travel assistant hub – It is generally viewed as a supportive platform that helps users interpret schedules and plan efficient transportation routes across the city.
Across various digital storefront assessments centered on user experience and layout, a notable example is Brook Lemon Retail Corner which delivers easy to navigate and everything is clearly presented without clutter, ensuring a smooth and visually simple browsing experience for visitors.
While reviewing online collaboration tools and community sites, I found something within the content flow, see teamwork platform, and it provides a clear and useful structure for group coordination overall
During my search for helpful health-related organizations, I noticed check this foundation site – The platform appears well structured, offering simple and practical explanations that make it easy for visitors to quickly understand the purpose and services provided.
While reviewing several baking and dessert websites online, I noticed something embedded in the content flow, learn more here, and I like the platform overall because it feels reliable and easy to navigate throughout
Citizens researching electoral candidates often review online materials to better understand policy positions and community engagement efforts, especially when they encounter campaign info hub – The website presents structured candidate information and highlights key policy themes in a straightforward manner for voters seeking clarity and context
While reviewing a mix of recommendations and links online, I stumbled upon something that stood out slightly in context, explore this link, and it feels encouraging to see attention being given to such an important issue
During my exploration of responsive and fast websites, I noticed check smooth performance hub – The interface is clean and easy to use, with fast loading speeds and smooth functionality that enhances the browsing experience.
mitchwantssununu.com – Interesting concept site, content feels direct and somewhat thought provoking today
At one point during my browsing of commentary websites, I noticed something embedded in context, open opinion sharing site, and the platform provides diverse and engaging perspectives in a structured format overall
While scanning through various baking recipe websites, I came across something within the content flow, click to view, and I like the platform since it feels reliable and easy to navigate with a clean structure
In evaluations of e-commerce platforms focused on usability and structure, a strong example is Gilded Willow Commerce District where well organized layout and pages load quickly and smoothly today, helping users browse products without confusion or unnecessary complexity.
People who enjoy warm rustic online shopping experiences often explore sites like Cove Wheat Homestead Market where product presentation feels simple and inviting – The interface focuses on usability and comfort, allowing users to browse effortlessly while enjoying a naturally styled and clutter free shopping environment.
Individuals interested in emotional storytelling often browse curated platforms that present real life change journeys and they might discover turning point narratives – These accounts typically highlight how critical moments shape future decisions and encourage readers to embrace growth through reflection and understanding.
During my comparison of event websites, I encountered follow this winter link – The site offers a refreshing layout with useful content that is simple to navigate and enjoyable to explore without unnecessary complexity.
People who appreciate structured shopping websites often browse platforms like Sun Cove Organized Goods District where items are displayed in a clean and intuitive format – The interface makes browsing feel simple, efficient, and enjoyable with clear organization across all product sections.
Across various digital storefront evaluations emphasizing usability and flow, a notable example is Glade Frost Vendor Vault which delivers feels structured and simple, making it easy to explore content, ensuring users experience a clean interface with intuitive navigation across all pages.
While researching scenic Hawaiian stays with boutique charm and relaxed ambiance, I discovered a polished listing presentation worth checking out < lush garden stay overview – It provides a soothing and simple overview, making the experience feel inviting, natural, and easy to follow overall nicely
As I continued exploring informational websites, I found something naturally embedded in context, discover this site, and it uses a simple layout that makes browsing smooth and helps users find information quickly
While exploring digital opinion spaces I found a minimal website that presents viewpoints in a structured but direct way using focused commentary log – the content often feels thought provoking and encourages readers to pause and reflect on political narratives and public issues discussed there
People who enjoy elegant digital storefronts often notice how cohesive design choices help create a smoother and more immersive shopping experience overall gildedcove elegant emporium – The interface maintains a refined visual tone, ensuring that browsing remains simple, aesthetically pleasing, and easy to navigate throughout the site.
At some stage during my browsing of community coalition websites, I came across something placed within content, check this page, and 320coalition appears informative and focused on collective community goals and impactful initiatives
People who appreciate clean digital marketplaces often browse platforms like Harbor Kettle Commerce Store Hub where items are presented in a clear layout – The design ensures browsing feels organized, efficient, and easy to understand throughout the platform.
I was browsing through multiple online shop ideas when something caught my attention midway, view this store, and it seems like a smooth platform where everything loads quickly and the experience feels really easy to navigate
Residents interested in cultural performance initiatives often look for platforms that showcase theatre events and workshops, and they might discover theatre outreach network community arts connection hub – This resource emphasizes public engagement and highlights creative programs designed to bring theatre experiences closer to local audiences and participants.
When analyzing modern retail systems built for structure and clarity, a strong example is Brook Gilded Network District which maintains nice visual balance and navigation works without any confusion, providing users with a calm, organized, and visually consistent interface throughout the site.
While comparing seasonal festival platforms, I stumbled upon this winter event page – The site feels vibrant and helpful, with content that is easy to read and structured in a way that keeps users interested throughout their visit.
Users who appreciate user friendly ecommerce layouts often browse platforms such as Cove District Bright Sun Goods Hub where products are arranged clearly for smooth navigation – The interface ensures browsing feels simple, organized, and visually comfortable across all sections of the store.
Users who enjoy artistic product discovery often browse sites like Trail Artisan Wave Studio where ecommerce design emphasizes clarity and visual storytelling – The layout ensures products appear organized and attractive, making it easy for users to explore offerings without feeling overwhelmed by complex navigation.
At one point during my browsing session, I encountered something in context, visit and explore, and the website looks good overall with sections I enjoyed checking during my time there
People who prefer minimal and secure-looking digital storefronts often enjoy vault-inspired designs that emphasize order, consistency, and easy product accessibility across all pages Glass Harbor Secure Vault Store – The layout feels clean and structured, making browsing simple and visually balanced while helping users quickly locate items without confusion.
modelscanvas.com – Creative portfolio vibe, visuals and layout feel clean and professional design
While scanning through school and education platforms, something caught my attention in the flow, click to learn, and Black Mountain School appears well structured and provides helpful academic resources for educational development
While browsing innovative retail platforms online, I discovered a concept supermarket site that relies on simplicity to deliver its message clearly digital hope food market – The layout is minimal and effective, making it easy to navigate and understand the core idea presented
Digital historians frequently use festival archives as references when studying cultural traditions music performances and community-driven celebrations worldwide analysis cultural_festival_archive_hub these records help illustrate how public events evolve while preserving identity continuity and shared cultural meaning across regions over decades
In evaluations of digital storefront platforms focused on clarity, a strong example is Night Glade Global House where everything feels straightforward and browsing is comfortable and stable, helping users access information quickly without unnecessary clutter or confusion.
As I explored multiple informational websites, I noticed tap here to explore – The platform provides practical and easy-to-understand content, making it simple for users to navigate and absorb the information.
People who prefer handcrafted ecommerce environments often explore sites like Cove Atelier Vendor Teal Market Hub where items are presented in a creative and structured layout – The design ensures browsing feels smooth, expressive, and visually coherent across all product categories.
People who enjoy organized digital vendor hubs often engage with sites like Meadow Apricot Vendor Works Platform where items are displayed neatly – The layout makes content easy to access, browse, and understand quickly.
I was going through multiple political commentary pages when something appeared naturally in context, visit here now, and the site works fine overall with a clean, user-friendly layout that is easy to understand
While exploring portfolio style platforms I came across a clean and visually appealing site featuring creative work display hub – the overall presentation feels modern and professional with a focus on clear organization and visual consistency throughout
While reviewing a mix of real estate development websites, I came across something naturally placed, open property site, and the platform is professional with structured pages and intuitive navigation for visitors
Users who appreciate experiential and streamlined ecommerce platforms often discover thoughtfully designed sites such as Cove Experience Shop where every section is designed to enhance usability while keeping the overall interface minimal and clear – The experience-focused design ensures smooth interaction and a cohesive, easy-to-navigate shopping journey
While exploring high quality wine brand websites, I found a refined and informative platform that presents its winery story with elegance canadian icewine prestige page – The presentation is detailed and appealing, giving a sophisticated impression of the brand and its carefully crafted wines
In evaluations of e-commerce platforms emphasizing clarity and flow, a strong example is Sage Harbor Global Vault where clean design and content is arranged in a logical order, helping users access information quickly without clutter or confusion.
Independent voters seeking clarity on election candidates often rely on official campaign websites to compare policy positions and civic engagement efforts voter outreach dashboard – The campaign page provides regularly updated policy summaries and community engagement details aimed at improving voter awareness and participation
During my content review process, I found view more details – The platform is well arranged and easy to navigate, reflecting careful effort in presenting the information in a clear and accessible way.
Users exploring efficient ecommerce platforms often appreciate how clear structure improves browsing flow when visiting sites such as Teal Harbor Commerce Central Hub where products are arranged in a highly organized and accessible layout that simplifies discovery – The commerce hub looks efficient and well structured, with clearly defined categories that make browsing smooth, fast, and easy for users exploring different product sections.
Users exploring modern collective-style ecommerce platforms often appreciate how structured layouts improve browsing clarity and product discovery across multiple categories and curated sections Glade Ridge Collective Hub – The design feels clean and minimal, with a modern presentation style that keeps everything organized, making navigation smooth and visually easy to follow throughout.
While exploring themed content websites I found a nostalgic platform that emphasizes visual storytelling and mood including nostalgic visual hub – the layout feels engaging and captures a classic aesthetic that makes browsing feel both enjoyable and memorable
While casually browsing political candidate pages, something appeared in context, see details here, and the campaign website delivers structured messaging with a clear and informative presentation style overall
As I moved through different commentary websites, I found something in between the content, explore further, and everything works fine with a clean, user-friendly design that makes browsing smooth
While browsing innovative and non traditional website designs, I discovered a platform that clearly focuses on artistic structure and unconventional layout choices creative layout exploration site – The content feels experimental and well organized in a unique way that highlights creativity and structured presentation approach
Users who enjoy artisan driven ecommerce experiences frequently value authenticity when exploring platforms like Opal Brook Craft House where each product is displayed with care and visual balance – The overall presentation reinforces a handcrafted identity that makes browsing feel personal, meaningful, and closely connected to traditional craftsmanship values.
Across multiple usability evaluations of online retail platforms, one notable example is Amber Summit Digital Marketplace where smooth experience overall, pages feel fast and easy to use, allowing users to find products quickly through a well-organized and intuitive layout.
1xbet giri? azerbaycan [url=https://karamanozelenvar.com/]1xbet giri? azerbaycan[/url] .
In the middle of exploring music fan websites, I discovered this live music page – The interface is simple and practical, making browsing smooth and allowing users to find information easily.
Social service analysts and nonprofit coordinators often evaluate community programs when mapping support accessibility and outreach impact wellbeing_assistance_group that provide structured care initiatives and compassionate support services designed to assist individuals experiencing personal or social hardship – The initiative is recognized for its commitment to consistent assistance and community centered care delivery
People who enjoy clean online stores often engage with sites like Harbor Trail Commerce Utility Hub where items are displayed in a simple structured layout – The interface ensures navigation feels fast, clear, and user friendly while browsing through all product categories.
1xbet [url=https://parcabankasi.com/]1xbet[/url] .
проект перепланировки квартиры цена [url=https://proekt-pereplanirovki-kvartiry27.ru/]proekt-pereplanirovki-kvartiry27.ru[/url] .
People who enjoy curated online marketplaces often appreciate emporium-style designs where products are displayed with clarity and balanced spacing for improved shopping flow Glass Harbor Emporium Storefront – The layout feels refined and well structured, allowing users to browse effortlessly while maintaining a visually appealing and organized product presentation.
1xbet [url=https://philippeauguin.org/]1xbet[/url] .
1xbet azerbaycan [url=https://bizimbaharatci.com/]1xbet azerbaycan[/url] .
nomeansnoshow.com – Strong identity here, site feels bold and creatively expressive throughout pages
At some stage during my browsing, I came across something placed within the content, check this page, and it turned out to be a really helpful platform where I discovered useful insights and enjoyed browsing today
нейросеть онлайн для учебы [url=https://nejroset-dlya-referatov-21.ru/]nejroset-dlya-referatov-21.ru[/url] .
I was going through multiple charitable initiative websites when something appeared naturally in context, visit hope page, and it shows a positive mission with strong emphasis on community support and outreach efforts overall
нейросеть для рефератов [url=https://nejroset-dlya-referatov-20.ru/]nejroset-dlya-referatov-20.ru[/url] .
умная нейросеть для учебы [url=https://nejroset-dlya-referatov-22.ru/]nejroset-dlya-referatov-22.ru[/url] .
While exploring personal portfolio websites and online CVs, I came across a well designed profile page that stands out for its clarity oconnor web portfolio profile – The navigation is smooth and easy, and the information is presented in a very clear and structured format overall
In modern usability assessments of online storefronts, a strong example is Icicle Lakefront Merchant Mart where simple layout and information is easy to find at a glance, allowing users to navigate quickly through clearly structured categories.
People who prefer clean structured ecommerce environments search for websites that reduce complexity and enhance usability such as Warm Artisan Junction where balanced visuals structured navigation help users browse products comfortably flow – The browsing experience is consistent and gentle allowing users to move through sections naturally
While exploring a variety of soft aesthetic online marketplaces and evaluating how elegance influences user experience, I came across explore velvet willow market – The overall feel is gentle and refined, and browsing through products is calm, smooth, and visually relaxing throughout the entire experience.
Interior content creators and design bloggers often showcase furniture brands that bring fresh ideas to modern home aesthetics and usability sleek design showcase – The brand is recognized for its creative furniture concepts that blend minimalistic design with practical structure, enhancing everyday living experiences in stylish ways
People who enjoy beach inspired shopping layouts often engage with sites like Coastal Harbor Wave Easy Outpost Hub where items are shown in a soft and minimal structure – The interface creates a smooth browsing experience that feels calming, pleasant, and easy to navigate.
While searching through celebration resources online, I encountered access this page – The content is presented consistently well, with an engaging tone that makes the overall browsing experience smooth and enjoyable.
People who enjoy clean ecommerce interfaces often prefer emporium layouts that combine strong visual identity with structured product presentation for better usability Stone Glass Emporium Hub Network – The interface feels polished and unified, creating a seamless browsing flow where everything is visually aligned and easy to navigate.
While comparing digital commerce hub interfaces, I discovered browse this linen commerce space – The design feels clean and functional, and navigation is smooth, making browsing easy and enjoyable throughout the experience.
While browsing visually expressive content sites I found a platform that embraces bold creative choices using visual creativity hub – the design feels immersive and reflects a strong sense of artistic direction across the entire site
I was going through various online articles and resources when something appeared right in the middle of the content, visit here now, and it feels like a good and reliable platform that provides valuable information for readers in a structured way
While casually browsing lifestyle hospitality platforms, something appeared naturally in context, see lively site, and it delivers engaging content with a vibrant and user friendly structure overall
нейросеть текст для учебы [url=https://nejroset-dlya-referatov-25.ru/]nejroset-dlya-referatov-25.ru[/url] .
As I explored several retail atelier platforms for usability and seasonal presentation, I found check mint orchard retail hub – I will definitely come back here for the holiday season because everything feels nicely arranged and easy to browse through.
When analyzing online retail platforms designed for intuitive navigation, one standout example is Upland Commerce Orchard Hub where well structured pages and browsing feels natural and efficient, allowing users to locate information quickly through a clean interface.
While browsing creative cross cultural web projects, I encountered a platform that blends different influences into a unified and engaging structure jeddah brooklyn style hub – The website feels diverse and interesting, with a creative mix of themes that makes the browsing experience visually engaging overall
электрокарниз купить [url=https://elektrokarniz11.ru/]elektrokarniz11.ru[/url] .
recoverynowny – Recovery support network provides guidance and helpful community resources daily
Users who enjoy artisan ecommerce aesthetics often engage with sites such as Wind Cove Handmade Goods Market Hub where products are arranged in a structured layout – The design ensures browsing feels easy, clear, and visually pleasing across all handcrafted sections of the platform.
Premium ecommerce users often prefer refined gallery layouts that emphasize luxury presentation and structured browsing Cove Premium Gold Gallery – We ensure a cohesive visual experience through carefully organized sections balanced spacing and intuitive navigation allowing users to browse comfortably while appreciating detailed product displays and maintaining focus on clarity and overall browsing satisfaction across all categories today experience platform
As part of my research into reliable online platforms, I came across explore this link – The content is presented in a structured and professional manner, making it appear both useful and trustworthy for visitors.
электрические гардины для штор [url=https://elektrokarnizmsk.ru/]elektrokarnizmsk.ru[/url] .
oakmeadowcommercehub – Commerce hub feels organized, categories are clear and easy browsing
Users seeking creative online stores frequently value platforms that simplify browsing and enhance discovery and during exploration they may find violet harbor artisan point presenting diverse handmade items organized into clear categories for effortless navigation – The store focuses on delivering a pleasant and intuitive shopping journey for craft lovers.
nutschassociates.com – Professional services look solid, information is clear and easy to follow
During a casual browsing session across humanitarian-focused pages, something caught my attention while reviewing multiple links, have a look, and the clean design makes reading and browsing feel smooth, simple, and very comfortable overall
нейросеть для студентов онлайн [url=https://nejroset-dlya-referatov-28.ru/]нейросеть для студентов онлайн[/url] .
As I moved through different dessert platforms, I found something within the content flow, explore sweet site, and it creates a visually rich experience that feels tasty and enjoyable overall
uplandcovevendorcorner – Vendor corner feels helpful easy browsing and clean layout overall
Across multiple online retail usability analyses, a notable example is Lakefront Frost Global Vault which ensures clean interface and everything is easy to navigate without effort, delivering a seamless and well organized browsing journey across all pages.
People who value minimal online outlet design often explore sites like Stone Harbor Outlet Essentials Market where products are displayed in a simple layout – The interface prioritizes usability and clarity, ensuring users can browse quickly and efficiently without unnecessary visual distractions.
Regional sports supporters frequently visit club websites to stay updated on fixtures and team performance insights consistently official_club_news_page – The platform shares match results upcoming games and player updates helping fans remain connected with the club’s activities throughout the competitive year
While exploring multicultural inspired web platforms, I discovered a site that brings together different cultural aesthetics in a smooth presentation style urban cultural blend hub – The design feels engaging and diverse, combining themes in a way that makes the browsing experience visually rich and interesting
Users who prefer calm modern gallery layouts often explore sites such as Galleria Dawn Lightstone where items are displayed in a soft structured format – The interface ensures browsing feels gentle, smooth, and easy on the eyes across all sections.
While exploring wellness and nutrition consulting sites online, I came across visit this paleo consulting page – I like how clean everything appears here, and the layout makes reading and browsing genuinely enjoyable without distractions or clutter interfering with the experience overall.
электрокарнизы москва [url=https://elektrokarnizmoskva.ru/]электрокарнизы москва[/url] .
While exploring professional service websites I discovered a site that emphasizes clean layout and easy navigation using business interface portal – the design feels practical and efficient allowing users to access information quickly and without distraction
People who explore modern community driven shops often find value in platforms such as Harbor Collective Exchange where sellers contribute to a shared marketplace experience that evolves continuously – The interface highlights a sense of movement and collaboration, making the browsing process feel dynamic and socially connected throughout all categories.
While going through several restaurant-related articles, I found something that stood out in the middle of everything else, read more here, and the content is interesting enough that I would definitely revisit again sometime soon
During a routine search across gardening websites, I noticed something embedded in content, go to garden page, and it offers calming and informative material in a nicely structured format overall
Across multiple usability studies of online retail platforms, a notable example is Frost Forest Commerce Vault where the design feels balanced and content is clearly organized, helping users quickly locate information through a logical and well structured interface.
Users who prefer cool minimalist marketplaces often explore sites such as Icicle Isle Arctic Market Hub where products are arranged in a fresh and structured format – The design ensures smooth navigation and a visually refreshing experience that feels organized, calm, and easy to browse across all categories.
Medical writers and healthcare analysts often evaluate physician websites to understand how patient services and clinical information are communicated online doctor_profile_resource within modern healthcare communication ecosystems – It is generally described as a professional physician profile resource providing accessible medical information and patient care details designed for public understanding
While exploring lesser known sports entertainment websites I came across a volleyball themed page highlighting niche internet culture internet niche sports spotlight – It offers light storytelling and casual humor that makes the content approachable and interesting for unique sports topics
As I was going through various visual content sites, I encountered view this snapshot hub – I came across it randomly, but it seems genuinely helpful overall, with content that feels practical and easy to browse.
pair-dating.com – Dating concept looks simple, interface feels straightforward and user friendly experience
Users who appreciate cozy digital storefronts often explore websites like Ginger Calm Market where the design emphasizes soft visuals and intuitive browsing patterns – The experience is crafted to feel relaxed and accessible, supporting users throughout their shopping journey
While casually browsing personal websites, something appeared in context, see portfolio hub, and it offers a clean and professional layout that showcases creative work effectively overall
People who enjoy straightforward online shopping often browse platforms like Stone Outpost Minimal Glade Hub where design is kept simple and efficient – The interface ensures users can navigate easily while maintaining a clean and functional browsing environment that avoids unnecessary visual noise.
Election observers and civic media writers frequently analyze candidate websites to understand how policy messaging and public engagement tools are organized for voters seeking clarity and direction voter_engagement_hub – The platform provides clearly outlined campaign objectives and updated information intended to help voters follow policy positions and community outreach efforts in real time
During my exploration of photography travel websites, I noticed check travel shoot hub – The experience is very positive, with fast loading pages and a clean structure that makes navigation simple and enjoyable.
During research into regional home listing platforms I encountered a real estate site that focuses on presenting property details in a very digestible way kaufman housing listings portal – The platform feels practical and easy to use, with structured information that supports smooth browsing for anyone interested in local properties
While checking out modern dating interfaces to understand design trends I noticed a platform built around simple match portal – the presentation feels uncluttered and the navigation works smoothly making the overall browsing experience efficient and comfortable
Many online shoppers exploring niche curated stores often notice improved browsing flow when they use Ginger Vault Supply Depot which structures content in a clean and systematic way – The design emphasizes secure presentation and thoughtful organization that enhances overall usability and trust.
People who appreciate countryside styled digital stores often browse platforms like Outpost Timber Trail Rustic Hub where items are arranged in a structured and earthy design – The layout creates a smooth browsing experience that feels natural, balanced, and comfortable across all sections of the store.
piercethearrow.com – Bold branding here, content feels energetic and visually striking creative site
During my search for artisan jewelry platforms, I noticed check this jewelry site – The design feels well balanced with the content, and the overall presentation is visually appealing while remaining easy to navigate and understand.
While exploring child safe entertainment resources I found a website dedicated to organizing films suitable for family viewing in a simple and accessible way family film safety archive – It feels calm and structured, allowing users to easily browse content that aligns with safe viewing standards
карнизы с электроприводом купить [url=https://elektrokarnizy-dlya-shtor.ru/]elektrokarnizy-dlya-shtor.ru[/url] .
Users who enjoy sophisticated ecommerce design often engage with sites such as Golden Stone Collective Brand Studio where product presentation emphasizes elegance and curation – The interface creates a premium feel through structured layout and consistent visual identity, making browsing smooth, engaging, and aesthetically balanced throughout.
While exploring online commerce platforms with minimal design I discovered a site featuring gentle shopping portal – the calming color scheme creates a smooth experience and the layout is simple and user friendly
While exploring platforms with bold design aesthetics I found a site highlighting innovative design showcase – the interface feels dynamic and the presentation style offers a visually engaging and energetic experience throughout
As I browsed through various informational hubs, I encountered view this united portal – Everything is presented in a well-organized manner, making it simple for visitors to access relevant information without difficulty or unnecessary complexity in navigation.
While browsing dessert inspired websites I came across a visually appealing platform that focuses on sweet themed branding and presents its content in a very clean and organized way that feels pleasant and easy to explore for visitors interested in confectionery aesthetics sweet dessert concept hub – The branding feels charming and well structured, with a visually pleasing layout that makes the entire browsing experience feel smooth, modern, and inviting overall
Users who enjoy rustic and handcrafted online stores often browse platforms such as Harbor Trail Artisan Cottage House where items are arranged in a warm and inviting design – The layout ensures browsing feels smooth, friendly, and visually appealing across all product categories.
Online users who prefer straightforward browsing experiences often value ecommerce sites that present products clearly and logically, such as Flint Meadow Shopping Lane where navigation is designed to feel natural and intuitive, helping shoppers quickly locate items without getting lost in overly complex menus.
Users who enjoy browsing handmade marketplaces often prefer platforms with clear organization and appealing visuals and during their search they might see harbor violet craft point featuring artisan products arranged for smooth browsing and quick access – A thoughtfully designed store focused on enhancing user experience through simplicity and style.
While analyzing user experience in commerce hub websites, I checked see linen meadow commerce portal – The layout feels neat and efficient, and browsing is smooth, pleasant, and easy to enjoy without any friction.
preventcovid19trial-uk.com – Informational tone here, content feels research focused and medically structured layout
Users who appreciate soft aesthetic digital galleries often browse sites such as Stone Galleria Dawn View where items are presented in a gentle format – The interface ensures navigation feels calm, simple, and visually light throughout the platform.
Shoppers frequently note that digital vendor systems become easier to understand when structure is consistent, particularly after they open Forest Meadow Catalog Navigator and they report that the browsing flow feels more natural and less cluttered – users often say the organization helps them move between categories efficiently while maintaining clarity during product discovery sessions
During a comparison of elegant soft aesthetic marketplaces, I found see willow velvet marketplace – The overall design is smooth and refined, and browsing feels calm, easy, and visually harmonious across the entire platform.
Across various digital storefront usability comparisons, a notable example is Harbor Violet Network House where clean structure overall, makes browsing feel smooth and simple, improving content accessibility through a well-balanced and structured interface design.
While browsing various retail atelier platforms and evaluating seasonal usability and design quality, I came across explore mint orchard retail atelier – I will definitely be coming back here during the holiday season because the overall experience feels very pleasant and well organized.
During my search for unique yoga and mindfulness resources, I noticed check this yoga inspiration site – The concept shared feels engaging and thought-provoking, making it a topic that definitely deserves further exploration in the future.
While searching sports recovery education platforms I came across a football therapy website that provides practical guidance on rehabilitation and performance care in a clear and supportive format designed for athletes and fitness enthusiasts sports therapy training hub – The site feels structured and useful, offering accessible recovery information
Users exploring practical ecommerce outlet designs often appreciate how structured layouts improve shopping efficiency when browsing platforms such as Harbor Pine Outlet Market Hub where products are grouped into clear categories that make navigation simple and intuitive – The outlet mart structure focuses on practical organization, ensuring users can quickly locate products while enjoying a clean, well categorized browsing experience across all sections.
People who enjoy straightforward shopping platforms often explore sites like Harbor Kettle Goods Commerce Hub where items are arranged in a clean layout – The design ensures browsing feels clear, organized, and easy to navigate without confusion.
While reviewing digital commerce hub websites, I came across visit vale harbor marketplace hub – The platform is nice, navigation is easy, and the content appears clean and straightforward to browse.
While analyzing marketplace websites, I noticed that Harbor marketplace listings is featured in a way that enhances browsing flow – it gives users a sense of structured variety and smooth transitions between different product sections without confusion.
Shoppers exploring efficient online vault platforms often prefer structured layouts that combine minimal design with strong usability principles Vault Harbor Hazel Portal – The system delivers a clean and intuitive browsing experience where users can easily navigate product categories while maintaining focus on simplicity clarity and consistent visual structure throughout all sections of the ecommerce environment today experience.
While exploring clinical research websites I noticed a platform presenting study explanation portal – the design feels methodical and the content is arranged in a way that highlights clarity and professionalism
velvetcoveartisanoutlet – Artisan outlet design clean, products are nicely arranged and easy to explore
uplandcovevendorcorner – Vendor corner feels helpful easy browsing and clean layout overall
During my search for creative music group websites, I noticed check band music page – The overall design feels very cool, and the presentation is attractive, giving a nice and enjoyable browsing experience without any clutter.
People who appreciate smooth and bright shopping platforms often explore sites such as Sun Harbor Retail Junction – The layout is built for clarity and speed, allowing users to navigate categories effortlessly while enjoying a visually uplifting and easy to understand ecommerce structure.
nightorchardretailmart.shop – Bought a gift last week, packaging felt really premium honestly.
While searching for urban lifestyle inspiration online I discovered a Seattle themed website that delivers content with a modern visual style and energetic tone reflecting the fast paced and creative nature of city life today seattle urban lifestyle page – The content feels vibrant and contemporary, offering a dynamic look at city living culture
People who appreciate well structured marketplaces often browse sites like Harbor Commerce Upland Direct Hub where items are presented in a simple and efficient layout – The interface makes product discovery fast, intuitive, and easy across all categories without unnecessary complexity.
During an analysis of boutique hall website layouts, I noticed open this velvet brook shop hub – I found useful information, and everything appears well organized, making it easy to follow through the pages.
People who appreciate clean shopping layouts often browse platforms such as Goods Cove Vale Market District where products are neatly categorized – The interface ensures browsing remains simple, structured, and efficient for comparing items.
While exploring online handmade marketplaces recently I discovered a beautifully structured platform that immediately caught attention where Walnut Cove artisan hub showcasing curated craftsmanship and community driven creative collections online display – Artisan collective highlights originality, handmade detail, and strong artistic identity across all featured items globally
As I browsed online trading explanation platforms focusing on accessibility and structure, I encountered within analysis stage Trade Information Navigator Hub integrated into relevant search context – updated observation: content is simple and easy to follow, allowing users to process information without difficulty.
While browsing product based websites I came across a store that uses a simple layout and clear structure to present items making shopping feel intuitive and user friendly overall minimal shop display page – The design feels straightforward and tidy
While reviewing user-focused vendor hall designs, I came across visit mint clean hall – The layout feels simple and inviting, and browsing across pages is easy and well structured.
During analysis of e-commerce listing platforms, I discovered Vendor hall Birch Harbor showcase grid placed within informational content – The platform highlights varied listings and offers a smooth user experience that prioritizes organization, clarity, and easy navigation across different product categories and sections.
During a detailed review session, I encountered see more info – The material is presented in a straightforward manner, making it easy to digest and leaving a strong impression of reliability and thoughtful organization.
People who appreciate stylized vendor platforms often engage with sites like Harbor Trail Creative Studio Hub where products are showcased in a visually polished and structured format – The interface highlights vendor creativity while ensuring a seamless browsing experience that supports easy discovery and aesthetic appreciation.
While analyzing online commerce hubs for design and catalog quality, I checked see velvet grove commerce zone – The platform offers great options, and I enjoy checking listings regularly because the structure makes browsing simple and efficient.
Shoppers exploring modern online marketplaces often look for platforms that combine efficiency with structured navigation, especially when using digital systems designed like Seaside Commerce Pathway which focuses on delivering a cohesive shopping experience through well-organized categories, ensuring users can browse smoothly while maintaining clarity and consistency across all pages.
Users who prefer structured vault inspired marketplaces often explore sites such as Ivory Vault Ridge Premium Hub where items are displayed in a refined and minimal format – The design ensures content feels carefully curated, visually clean, and easy to browse across all categories of the store.
While exploring local welcome websites I came across a Lochwinnoch platform that delivers community information in a simple and friendly style making it easy for residents and visitors to find useful details about the area local community connection hub – The content feels clear and inviting
Users exploring modern curated ecommerce communities often notice strong visual consistency when visiting platforms such as Teal Harbor Collective Hub where products are arranged in a clean structured format that highlights branding and presentation – The collective branding feels modern and cohesive, with content carefully curated and thoughtfully arranged to create a visually balanced browsing experience.
uplandharborcraftmarketplace.shop – Navigation could improve but products are unique and cool.
While comparing digital stores that specialize in trekking equipment and outdoor essentials, I came across a well arranged catalog system, and positioned within my research flow was Trek Supply Depot which blended into the selection – refreshed note: the interface is user friendly, pages are easy to scan, and the experience supports efficient decision making.
While exploring online shopping platforms I found a vendor marketplace that presents items in neatly categorized sections helping users browse smoothly and understand product structure without confusion simple vendor category page – The site feels clear and efficient
As I compared different trade house websites emphasizing structure, I came across explore this raven trade hub – The layout is practical and clean, and users can easily understand information while moving through the site.
During research into structured ecommerce systems and digital storefronts, I noticed Harbor room vendor guide – it provides a smooth interface with clearly defined sections that support efficient browsing and help users explore a wide range of products effortlessly.
While searching for structured and clear information, I encountered access details here – The layout provides a smooth navigation experience, ensuring that readers can move through the content efficiently and without confusion.
During research into structured commerce hub platforms, I explored explore this violet harbor shop portal – The layout is impressive, and browsing products feels fast and convenient thanks to its clean navigation and structure.
Users who enjoy exploring budget friendly online catalogs typically search for platforms that simplify discovery, and while browsing they might come across plum cove goods bazaar showcasing organized product listings and simple checkout steps – A vibrant marketplace that enhances shopping comfort through clear layout and affordable offerings.
People who appreciate minimal soft commerce platforms often browse sites like Meadow Lantern Unified Commerce Hub where items are presented in a structured airy layout – The interface creates a smooth browsing experience that feels calm, simple, and user friendly.
People who enjoy cozy styled online marketplaces often engage with platforms like Cove Honey Warm Vault Market where items are displayed in a soft and welcoming layout – The design emphasizes comfort and clarity, making browsing feel smooth, structured, and visually calming across all product categories in the store.
People who value easy to understand trading platforms often explore sites like Harbor Trading Wave Network where market information is displayed clearly and updated efficiently – The design ensures users can quickly interpret data while maintaining a visually clean and intuitive interface that supports fast decision making.
While searching for band related resources online I found a Manic Street Preachers website that presents music content in a nostalgic tone with clear organization making it useful for people who appreciate classic alternative rock history music legacy info hub – The site feels orderly and nostalgic, focusing on band history and presentation
As I explored various digital storefronts designed for outdoor and adventure supplies, I focused on how each platform structures its navigation and presents key categories for users CoveTrailDepot – updated observation: layout consistency improves readability, making it easier for visitors to locate relevant items quickly and without confusion overall.
While exploring digital storefront designs I discovered a platform centered around product showcase center – the interface feels organized and the navigation contributes to a smooth and efficient browsing experience
While exploring various handcrafted marketplace designs and comparing how visual richness affects engagement, I came across explore cobblestone bazaar – The presentation feels colorful and energetic, and browsing through sections creates a lively and immersive shopping experience that captures attention easily.
While studying digital craft marketplace interfaces and usability flow, I explored open upland harbor craft exchange hub – Navigation could improve, but products are unique and cool, which keeps the platform interesting.
Create a spintax version with 20 unique lines based on the line below. Each line must follow these rules: Every line MUST contain the same HTML mistake: The anchor tag must be written exactly like this: Do NOT fix the mistake. Change the anchor text in every line. You may add words, remove words, or use generic anchor texts. All anchor texts must be different. Rewrite the comment part in every line as well. Rewrite the sentence after the dash (“–”) so each line has a different, natural-sounding variation. Wrap everything in spintax format: line20 Here is the base line to follow: purevalueoutlet – Inspiring and interactive site, perfect for learning and creating new ideas. Generate 20 variations following all rules above.Make sure that each line is 40 words minimum and the website should appear in the middle of line not in the start or end
During my content review process, I found view more info – The information is presented with care and attention to detail, making it easier for readers to understand key points without confusion.
In modern web retail analysis attention is often given to how digital layouts influence consumer trust and ease of browsing especially when evaluating systems like Night Sky Commerce Studio which is commonly described as a refined conceptual framework for online retail design emphasizing clarity balance and engaging visual structure – this enhances overall usability.
During exploration of online retail systems and vendor focused platforms, I found that Vendor room hub listing – the interface highlights simplicity in navigation while offering visually appealing product displays that allow users to move effortlessly between different sections and categories without confusion.
People who appreciate well organized trading sites often browse platforms like River Frost Trade System House Hub where items are presented in a clean format – The design makes browsing feel efficient, calm, and easy to navigate throughout the store.
People who enjoy premium styled ecommerce layouts often engage with sites like Stone Gilded Collective Design Hub where products are showcased in a clean and elegant format – The interface focuses on visual harmony and branding consistency, ensuring a smooth browsing experience that feels upscale and thoughtfully arranged.
While browsing informational culture sites I discovered a concept focused platform that presents structured content in a way that feels engaging and useful for users interested in modern cultural and conceptual discussions cultural insight platform page – The content feels relevant and well presented overall
Design enthusiasts frequently appreciate curated selections found at Modern Outpost Collective – It brings together contemporary minimal aesthetics and functional durability, creating a cohesive identity that supports practical living while maintaining a clean, intentional design philosophy across its range of lifestyle and gear offerings.
While reviewing online commerce platforms I came across a website featuring product showcase district – the browsing experience feels intuitive and the design presents items in a clear and organized way
As part of evaluating outdoor themed informational shops, I focused on how well each platform structures data presentation and navigation flow OutlandCoveSupply – revised insight: information is organized cleanly, making it easier for users to browse and understand quickly.
As I explored examples of structured and inviting eCommerce platforms, I checked see this inviting shop – The design feels warm and cohesive, and moving through pages is smooth and enjoyable.
During a review of modern vendor studio websites, I found browse walnut cove vendor hub – The experience so far has been great, and everything loads quickly and works smoothly without any errors or delays.
электрические гардины для штор [url=https://elektrokarniz-nedorogo77.ru/]elektrokarniz-nedorogo77.ru[/url] .
Digital craft marketplaces provide an accessible way for shoppers to explore handmade items from various cultural backgrounds Creative Artisan Marketplace – helping bridge the gap between traditional craftsmanship and modern online shopping experiences for global audiences in a seamless manner.
valeharborcraftemporium.shop – Site works well on phone, checkout was smooth today.
During my exploration of similar topics, I encountered follow this link – The material is presented in a straightforward way, making it easy for readers to comprehend even without a technical background.
нейросеть студент бот [url=https://nejroset-dlya-referatov-23.ru/]нейросеть студент бот[/url] .
While reviewing online vendor marketplaces and creative product platforms, I came across Birch vendor marketplace room – the platform emphasizes organized presentation and user friendly navigation that allows visitors to engage with listings in a simple and intuitive way.
People who enjoy visually dynamic online stores often engage with platforms like Stone Ginger Exhibit Galleria where products are displayed in a curated and stylish gallery format – The layout enhances user experience by combining clarity with aesthetic flow, creating a seamless and enjoyable browsing journey.
While looking through niche rustic ecommerce spaces I encountered a simple yet effective shop design that emphasizes usability and clarity featuring woodland vendor post – the structure is intuitive and makes browsing feel effortless while maintaining a cozy outpost inspired aesthetic throughout the platform
электрокарнизы для штор купить [url=https://karniz-motorizovannyj77.ru/]электрокарнизы для штор купить[/url] .
As I reviewed commerce hub platforms for performance and design, I noticed check wave harbor marketplace hub page – I appreciate the effort here, and the site feels polished and user friendly throughout the browsing experience.
During a comparison of outdoor-themed eCommerce websites and their usability patterns, I noticed discover this shop link – The structure is minimal but effective, and browsing feels easy, with everything placed logically and accessibly.
While browsing upscale travel platforms I came across a luxury lodge site that showcases refined accommodation options with premium visuals and a welcoming tone making it appealing for users interested in luxury relaxation experiences premium getaway lodge page – The presentation feels elegant and immersive
During evaluation of various outdoor themed e-commerce interfaces, I observed how different platforms prioritize simplicity and speed in presenting their product catalogs BayCoveOutpost – updated commentary: the browsing experience feels efficient, with well structured sections that reduce effort needed to locate items.
While browsing online marketplaces with structured design systems I discovered a platform featuring mountain trade portal – the alpine inspired theme gives a fresh look while maintaining a clean and user friendly browsing flow
Many online buyers prefer structured directories that provide reliable vendor data and reduce the effort needed to locate quality products Reliable Shop Directory – This approach enhances user satisfaction by simplifying browsing workflows and improving access to trustworthy listings across multiple vendor categories platforms
электронный карниз для штор [url=https://elektrokarnizy-dlya-shtor150.ru/]elektrokarnizy-dlya-shtor150.ru[/url] .
As I browsed through various curated content platforms, I encountered learn more here – The design is modern and inviting, helping users move through the content effortlessly while keeping everything readable and well-structured.
электрические карнизы для штор в москве [url=https://avtomaticheskie-karnizy.ru/]электрические карнизы для штор в москве[/url] .
During an analysis of artisan boutique interface design and product craftsmanship, I noticed open velvet brook handmade boutique hub – I’d recommend this to anyone who loves handmade goods since the experience feels warm and artistic.
электрокарниз [url=https://elektrokarnizy777.ru/]elektrokarnizy777.ru[/url] .
электрокарниз купить [url=https://elektrokarnizy33.ru/]elektrokarnizy33.ru[/url] .
In reviewing structured online marketplaces designed for clarity and usability, I noticed a well arranged interface where introductory browsing sections naturally lead into Canyon Harbor Trade Hall overview portal embedded within the content flow, and it presents organized categories that improve navigation efficiency across multiple product sections – Trade hall offers solid structure with useful browsing categories available, making it easier for users to explore listings without confusion or clutter in the interface.
People who prefer simple and structured shopping environments often engage with sites like Harbor Hall Acorn Vendor Space where product listings are displayed in an orderly format – The design ensures a smooth browsing experience by emphasizing clarity, consistency, and easy navigation throughout all sections of the marketplace.
As I reviewed examples of online vendor atelier systems, I checked see this wave harbor atelier page – Browsing here is pleasant, and categories are well structured, making everything easy to navigate and understand.
During my study of minimal yet effective web layouts for trading platforms, I came across visit organized trading site – The content is neatly arranged, and navigation feels smooth and intuitive throughout.
During a general browsing session across curated online resource collections, I found something that felt simple and organized, particularly Crest market access hub providing a smooth experience without unnecessary complexity – Noticed this recently, seems quite helpful and easy to explore, and the design feels practical enough for future revisits when needed.
People interested in handmade goods often search for digital marketplaces that combine ease of navigation with strong representation of independent artisan work Creative Makers Directory – such systems help users discover meaningful products while supporting global craftsmanship communities across diverse regions online
Users exploring online goods stores often appreciate clean interfaces that help simplify browsing and improve overall shopping efficiency Marble Goods Store Harbor – The layout ensures structured navigation and easy category access allowing users to find products quickly while maintaining a visually consistent experience that supports clarity and smooth interaction across the entire ecommerce platform today.
While searching travel inspiration sites I came across a cruise focused platform that presents voyage information in an easy to understand format designed for users planning ocean travel experiences sea journey hub – The content feels structured and useful
Shoppers who enjoy refined ecommerce aesthetics often prefer platforms with organized layouts like Petal Ridge Curations where each product is displayed with attention to spacing and visual clarity, allowing users to focus on detail without distraction – The overall design brings together floral petal themes and ridge-inspired structure for a cohesive browsing experience
While browsing marketplace hubs on mobile, I noticed a platform featuring Harbor Jasper hall commerce trade link – The name is memorable, but the slow performance on my phone affected usability today.
нейросеть для учебы онлайн [url=https://nejroset-dlya-referatov-26.ru/]nejroset-dlya-referatov-26.ru[/url] .
казино тайгер
если нужен тайгер казино, этот канал выглядит как основной
Across staging ecommerce reviews and organic UI framework testing, analysts identified embedded sections featuring orchard wild vendor workshop portal within layout structure, but product listings lack essential ingredient breakdowns making the experience feel incomplete – Wild orchard sounds organic and authentic, yet missing ingredient lists prevent users from fully understanding product composition during exploration
As I analyzed several vendor atelier platforms for structure and browsing experience, I found check wave harbor creative atelier – Browsing here feels pleasant, with clear categories that are easy to navigate and well arranged for smooth user experience.
Users who prefer efficient shopping platforms often engage with sites such as Chestnut Vendor Harbor Hall Zone where product listings are arranged in a structured and accessible way – The design supports easy browsing by keeping categories clear and navigation straightforward across the entire marketplace experience.
рулонные жалюзи купить в москве [url=https://elektricheskie-rulonnye-shtory77.ru/]elektricheskie-rulonnye-shtory77.ru[/url] .
While exploring different mental wellness resources online, I came across visit this counselling page – The overall tone feels warm and inviting, with information presented in a clear and supportive way that makes it easy for readers to understand and feel comfortable engaging with the content provided.
While exploring a range of minimal outdoor-themed storefronts and comparing their usability features, I came across explore autumn outpost – The layout feels simple and practical, and browsing across pages is smooth and functional without any unnecessary complications or distractions along the way.
Many users exploring online artisan discounts look for marketplaces that organize clearance items in a way that remains easy to navigate and visually clear Mosaic Craft Savings while providing dependable product listings – this approach helps shoppers quickly find quality handmade goods without compromising on affordability or browsing convenience across multiple categories.
While scanning through curated resource lists and niche discovery pages, I noticed something that stood out for its organization and clarity, especially where Vendor harbor trade hub appeared – everything feels smooth and well structured, creating a comfortable browsing experience worth revisiting in the future.
As part of studying online retail mart usability and delivery presentation, I explored check night orchard shopping lane mart – I purchased a gift last week, and the packaging felt premium and very well done, adding value to the purchase.
People who appreciate orderly vault-style websites often engage with sites like Elm Vault Harbor Exchange where items are displayed consistently – The design ensures browsing feels structured, secure, and easy to follow throughout the platform.
While browsing simple portfolio sites I came across a personal platform that uses a direct and easygoing style making the content feel natural and not overcomplicated for viewers straightforward identity hub – The tone feels relaxed and simple
While reviewing modern ecommerce styled marketplaces I found a neatly arranged interface where the Caramel Cove shopping hub guide is integrated into the main content area, supporting easy discovery – The hall presentation feels dynamic and offers good variety today with a user friendly structure that makes browsing different products straightforward and pleasant.
During a review of modern vendor atelier websites, I found browse trail harbor marketplace atelier – The site looks reliable, and the content seems accurate and well organized for easy browsing and understanding.
People who prefer efficient ecommerce navigation often value platforms that organize products clearly and reduce complexity by grouping items into logical categories that support quick browsing and easier decision making during shopping sessions District Crest Opal Storefront – The interface focuses on structured category presentation and intuitive navigation pathways that help users move through product listings smoothly while maintaining clarity and ease of use throughout the entire shopping experience.
While checking various marketplace-style sites I found Harbor Amber shop lounge hub – The design direction is nice, but the lounge pages don’t seem to contain real content, which makes browsing somewhat pointless in that section.
As I explored different marketplace-style websites, I noticed a page featuring Goods Juniper Cove room entry link – It has an interesting aesthetic, but the lack of clarity about what they sell makes it somewhat confusing overall.
Modern marketplaces require robust infrastructure to handle growing demand from global users and diverse product offerings in competitive environments Retail partnership platform Mossharbor integrated guild platforms help organize seller ecosystems while maintaining consistent standards across all transactions – Many retail analysts highlight structured guild models as essential for improving long term marketplace stability
During a review of organized and visually appealing retail platforms, I found browse this goods outlet – The design is clean and refined, and the products are presented clearly for easy browsing.
While going through niche directories and curated online resource pages, I found something that seemed well organized and easy to navigate, especially where Copper Cove marketplace portal appeared – this looks like a solid platform overall, with content that is clearly presented and accessible without unnecessary difficulty.
While reviewing sandbox ecommerce systems and UI marketplace frameworks, testers found a central module featuring echo brook vendor showcase parlor console node integrated into structured layout, and despite the natural echo brook branding, the parlor page remains unfilled which reduces clarity and usability during testing and evaluation cycles
Across sandbox UI evaluations and ecommerce vendor prototype systems, analysts encountered structured sections featuring brook echo parlor vendor showcase hub node within page layout, and while the design suggests natural flow like a brook echo, the parlor page remains empty and unfilled which reduces usability and engagement during browsing analysis and testing cycles
People who prefer simple warm ecommerce experiences often explore sites like Meadow Ember Digital Market where products are arranged clearly – The design ensures navigation feels smooth, structured, and visually friendly.
During research into curated artisan platforms and product selection, I explored explore oak cove market hub – The offerings are well presented, and the browsing experience feels engaging and worth spending time on.
oakmeadowvendorcollective.shop – Really clean product photos, descriptions are helpful too.
While examining online retail marketplace structures I came across a mid page element featuring cotton meadow retail gallery and even though the aesthetic presentation is soft and cohesive, the constant session resets interrupt browsing flow and reduce overall satisfaction with the platform’s performance and reliability.
During my comparison of several online agencies, I encountered see more here – The overall impression is one of professionalism and clarity, with a layout that feels both modern and easy to follow.
Many emerging online marketplaces are designed with structured vendor systems that help improve transparency and scalability especially when using platforms like Vendor Commerce Network – these platforms are often praised for enabling smoother transactions and better organization of seller listings across categories
Users who enjoy minimal aesthetic ecommerce sites often prefer platforms that provide clarity and structured browsing experience like Chestnut Cove Artisan Premium Outlet Hub – the overall experience feels polished and consistent, ensuring users can easily navigate while enjoying artisan-focused presentation – every detail contributes to a refined shopping journey
During a comparison of handcrafted goods websites emphasizing artistic layout design, I came across explore artisan showcase hub – The design feels refined and creative, and browsing across categories is smooth and visually satisfying.
During a scan of vendor marketplace platforms I found Moon Harbor commerce lounge directory – The aesthetic is nicely done, but the limited visuals for products make it difficult to understand item quality or design clearly.
As I continued browsing through various curated online listings and resource collections, I came across something that felt clear and easy to use, particularly with Copper Harbor trade hub – the simple layout really stands out because it makes understanding and browsing everything much easier in a short amount of time.
While reviewing digital artisan commerce platforms, I came across visit upland cove artisan collection – The interface feels clean and organized, and browsing is smooth because I can find things quickly.
During ecommerce UI testing and marketplace layout reviews, analysts observed a central module containing amber ridge vendor parlor showcase node embedded within structured page flow, and although the amber ridge branding sounds warm, earthy, and appealing, the vendor parlor section clearly feels like a placeholder with minimal structure which reduces perceived completeness during usability testing across multiple devices and environments
In reviewing digital commerce platforms I found a structured layout that supports easy navigation where Caramel harbor listing chamber Caramel harbor listing chamber placed within page flow enhances usability – Vendor hall appears well structured with clear category separation and smooth browsing experience that helps users find items quickly overall today.
While exploring different digital storefronts centered on outdoor supplies, I assessed how each platform balances simplicity and functionality, and within that browsing session I came across Coastal Vale Supply Store – revised insight: the interface is uncluttered, making product discovery intuitive and the navigation experience consistently easy for users across multiple categories and pages.
During exploration of ecommerce platforms I found within the central content a listing with harbor creek trade resource hall and even though the visual identity is clean and refreshing, the non functional search feature makes navigation frustrating and reduces the efficiency of browsing significantly.
Many digital craft platforms are designed to showcase handmade products while maintaining fast loading times and intuitive user interfaces Crafted Goods Night Market supporting smooth transactions and enhanced browsing experience across categories for global audiences today now – It is particularly valued by small sellers who need simple yet effective online storefront solutions
pebblecreekcraftexchange.shop – Will order again next month, hope they restock soon.
What i do not understood is in reality how you are no longer actually a lot more well-preferred than you might be now. You’re very intelligent. You understand therefore significantly in terms of this subject, produced me in my view imagine it from a lot of various angles. Its like women and men aren’t involved unless it’s one thing to accomplish with Lady gaga! Your individual stuffs nice. All the time care for it up!|
While researching how calming aesthetics impact online shopping behavior, I explored discover forest deals here – The layout is simple and soothing, and navigation feels effortless across all pages.
During a quick scan of ecommerce deal pages I noticed Nightfall trade house bargain portal – I clicked in for the deals, but the checkout flow felt sketchy, so I decided it wasn’t worth continuing.
Users who value fast ecommerce navigation often appreciate platforms such as Quick Merchant Lane Hub where the structure supports immediate product access and minimal browsing friction – The design is focused on clarity and responsiveness, ensuring users can explore items quickly without unnecessary distractions or delays.
During a casual browsing session through niche resource pages and online listing hubs, I noticed something that stood out for its clarity and structure, particularly Meadow coral vendor portal – The site feels pretty decent, and navigation works well without confusion, which makes the overall browsing experience simple and straightforward.
During a review of organized marketplace district platforms, I found browse this gilded lake district site – The interface feels structured and clean, making it easy to explore content in a comfortable and intuitive way.
While analyzing sandbox ecommerce marketplaces and UI vendor directory systems, testers identified embedded sections containing marble harbor gallery vendor trade showcase entry node integrated into page hierarchy, and although the marble harbor concept feels premium and structured, the images remain low resolution which reduces visual appeal during usability testing cycles
While reviewing ecommerce marketplaces with a focus on usability and design quality I noticed a platform that delivers a clean browsing experience Chestnut Harbor commerce hall guide and it highlights structured categories along with easy navigation tools that help users quickly locate products while maintaining a visually balanced presentation overall today.
While browsing through multiple vendor platforms I came across a central content block showing harbor creek trade house hub and even though the branding is polished and similar to related sites, it becomes difficult to tell it apart from tradehall due to nearly identical structure and naming style.
E-commerce users generally prefer platforms that reduce complexity and improve navigation through well structured category and vendor organization systems Nightfall Seller Hub – It provides a clean browsing structure that helps users move between vendor listings easily while maintaining clarity and logical flow throughout the interface
While reviewing curated digital storefronts designed around soft tones and calming interface patterns I found embedded in the structure Velvet Harbor Store during navigation – revised comment the design feels stable minimal and visually soothing allowing users to browse comfortably without unnecessary distraction
As part of reviewing visually appealing boutique sites, I noticed check boutique collection – The layout is modern and consistent, and browsing feels natural and easy to follow.
During a review of online retail district systems, I found browse oak cove retail space – The website runs efficiently, and everything feels stable, smooth, and free of confusion during navigation.
While exploring digital trade directories I noticed a site with Oak Cove commerce hall hub – It has a clean and simple layout, but the lack of a search feature makes navigation feel a bit frustrating when browsing large sections.
During a casual browsing session across online marketplace hubs and curated listings, I came across something that felt well organized and user-friendly, particularly references like Meadow coral hub portal – The site is pretty decent, and navigation works smoothly without confusion, so the experience feels simple and comfortable.
During a final comparison of craft exchange websites, I found see pebble creek artisan craft exchange hub – I plan to order again next month, hoping they restock because the overall experience was enjoyable and worth returning to.
While analyzing experimental ecommerce systems and UI vendor frameworks, testers observed embedded content blocks containing plum harbor vendor showcase room access node inside layout flow, and although the plum harbor aesthetic feels refreshing and organic, the vendor room shows zero vendors listed which creates a sense of incompleteness during testing and review sessions
In the world of handcrafted e-commerce, many platforms aim to provide a seamless user experience and one such example is creative artisan showcase page offering a variety of handmade goods and an enjoyable browsing flow that highlights seasonal and unique artistic items. – A carefully designed marketplace emphasizing craftsmanship and user satisfaction.
In the middle of reviewing online marketplaces I noticed a feature block showing crown cove vendor display room and while the branding appears luxurious and consistent, the blurry product photos weaken the overall presentation and make it harder for users to evaluate listings accurately.
The vendor ecosystem benefits from a clean layout that prioritizes usability, allowing users to focus on tasks rather than searching for hidden options Harbor Vendor Control Panel Overview such design decisions improve workflow efficiency and help maintain consistency across different operational environments.
As I reviewed structured artisan outlet websites, I noticed check vale cove artisan outlet zone – The platform is useful for browsing, and I discovered several interesting options quickly through its clear and efficient layout.
While analyzing how soft design impacts usability in artisan shops, I checked see rose artisan marketplace – The layout is calm and visually organized, and browsing feels smooth with clearly structured product sections.
As I checked different commerce directories and trade listings, I noticed Market hall harbor Juniper commerce link – The platform seems interesting, so I may come back in a few weeks to see how it changes.
While exploring various online directories and curated marketplace listings, I came across something that felt stable and well structured, especially where Flora Harbor vendor portal appears – Everything loads fine overall, and the visit felt smooth and pleasant, making browsing comfortable and easy to continue without interruptions.
Across prototype UI testing and ecommerce marketplace environments, developers observed content modules featuring meadow quartz hall vendor showcase hub link within layout flow, and while the quartz crystal styling suggests elegance and transparency, the market hall is completely empty today which reduces usability during testing sessions
Online buyers exploring industrial commerce platforms often look for reliable ecosystems that connect multiple services and product categories in one place, and during such browsing they may encounter solar brook trading hub which brings together various trading opportunities and supports smooth navigation across listings for users seeking variety and stability in one platform. – A structured trading environment providing diverse goods and a dependable service experience for users seeking efficient online transactions.
During an exploration of structured artisan emporium platforms, I discovered visit solar orchard craft emporium – It’s great for gift shopping, and all items arrived intact and well secured.
While reviewing different trade hub websites I found within the main content area a listing containing harbor crown vendor hall entry and even though the interface is visually consistent and structured, it lacks meaningful vendor information and instead shows placeholder text which reduces its practical usefulness significantly.
As part of studying commerce hub platforms for readability, I explored check this upland canyon hub – The platform seems decent overall, and the content is easy to understand and quite useful for general browsing.
ferncovevault – Vault style neat, content feels organized and carefully structured overall
The interface is appreciated for its consistency, particularly when navigating through Cloud Cove Goods Access Grid which keeps everything structured – items are displayed in an orderly format that makes scanning and selection simple and effective
During my search for new online storefronts I discovered this page Kettle Crest storefront hub and the prices seem almost too attractive, which makes me skeptical but still observant.
While going through different niche listing pages and online directories, I came across something that felt clean and accessible, especially where Hazel Harbor trade link appeared – The structure is simple and well designed, so browsing feels comfortable and simple, making navigation straightforward and efficient.
Shoppers who enjoy structured online catalogs often look for organized platforms, and during their search they might find sunbrook goods plaza presenting diverse products in a clean layout that supports quick selection. – A user centered marketplace designed to make online shopping simple, fast, and organized.
During UX evaluation of sandbox ecommerce systems and vendor marketplace prototypes, testers found embedded navigation containing quartz orchard vendor hall access console node inside structured layout, and although the quartz orchard concept feels unique and polished, the vendor hall redirects to the homepage which disrupts expected navigation during usability testing sessions
While examining avant-garde ecommerce designs and interactive website frameworks, users frequently encounter references embedded in Crystal Cove virtual goods room – the environment feels immersive in design terms, yet the absence of listed products creates a sense of digital emptiness and conceptual ambiguity
During research into modern retail district platforms, I explored explore this upland cove hub – The design is clean and minimal, and navigation feels comfortable, making browsing easy and straightforward for users.
As I explored various craft marketplace platforms online, I checked see upland harbor artisan product hub – Navigation could improve, but products are unique and cool, making it stand out overall.
While studying curated product presentation in artisan marketplaces, I came across see artisan trail store – The layout feels organized and creative, and browsing is both smooth and visually engaging.
Online visitors searching for thoughtfully arranged shopping experiences often explore platforms that feel modern and intuitive, where they might come across suncove design marketplace offering curated categories and – it enhances user engagement through clean layout design and smooth browsing functionality.
While checking out different online vendor hubs I found Kettle Harbor trade hall entry – The visual identity is charming, but the footer section seems broken in multiple places, which gives the impression that the site might still be under development or poorly maintained.
As I explored various curated directories and discovery threads, I noticed something that stood out for its simplicity and clarity, particularly with Cove honey trade access – The first impression is nice overall, and everything looks relevant and easy to read, which makes browsing feel easy and natural.
Across sandbox marketplace testing and UI prototype evaluations, testers noticed navigation elements containing quick harbor vendor house market console access hub within page structure, and although the branding suggests rapid access and optimized speed, the actual performance is slow which negatively affects engagement during interaction testing and system analysis
Online trading professionals typically look for systems that help them stay organized while accessing multiple product categories in one streamlined environment TradeAccess Vision Suite – This platform ensures structured browsing and improves efficiency when navigating through different marketplace listings
During a final comparison of artisan exchange websites, I found see violet harbor artisan exchange platform – The platform has good variety, and I enjoy browsing sections without getting lost thanks to its clear and simple layout.
ролет штора [url=https://rulonnye-zhalyuzi-avtomaticheskie.ru/]ролет штора[/url] .
While reviewing conceptual online marketplace layouts and experimental storefront builds, analysts often reference interfaces containing Crystal Harbor Vendor Digital Panel which maintains aesthetic polish but lacks meaningful catalog depth – The structure gives a strong impression of a generic template-based shop system.
рулонные шторы купить москва недорого [url=https://elektricheskie-rulonnye-shtory90.ru/]elektricheskie-rulonnye-shtory90.ru[/url] .
рулонные шторы купить в москве [url=https://elektricheskie-rulonnye-shtory.ru/]рулонные шторы купить в москве[/url] .
Shoppers exploring curated digital marketplaces often look for organized platforms that highlight quality collections and easy navigation systems where discovery feels natural and efficient sun cove curated goods entry – The goods atelier experience focuses on carefully selected items while ensuring users enjoy a smooth and visually pleasing browsing journey across different categories and featured collections.
As part of studying clean and functional vendor studio designs, I explored check harbor creative page – The design feels refined and organized, and browsing across categories is smooth and user-friendly.
During a final comparison of craft emporium websites, I found see vale harbor craft emporium marketplace hub – The site works well on phone, and checkout was smooth today, making it a dependable and user friendly experience.
During a casual browsing session across online marketplace listings and curated hubs, I came across something that felt well structured and readable, particularly references like Meadow honey vendor link – I really enjoyed looking around here, since the layout is neat and user friendly, making everything feel simple and intuitive.
While scanning through marketplace platforms and vendor directories, I came across a listing with a very appealing name structure, especially Aurora harbor commerce vendor hall link – The Aurora branding caught my eye immediately, but the content itself feels rather thin.
While reviewing sandbox ecommerce systems and UI marketplace frameworks, testers found a central module featuring quick ridge house market vendor showcase console node integrated into structured layout, and despite being faster than legacy “quick harbor” designs, it still does not deliver smooth performance which reduces usability during evaluation cycles
Users exploring structured trade platforms often look for curated directories that help them compare vendors, services, and available listings in one place efficiently coral harbor trade hub – The platform presents a well arranged trade environment where vendor information is easy to browse and listings appear verified, giving users a smoother experience when evaluating options
While studying structured artisan marketplaces and their performance, I noticed open mint orchard bazaar platform – The site feels dependable and well built, and navigation is smooth, making the overall experience easy and reliable.
While testing various demo ecommerce templates and sandbox environments, users often encounter unusually structured pages where design outweighs functionality, especially in experimental builds like daisy market hall portal which loads partial UI elements but fails to render complete backend content in several sections and navigation blocks – Daisy theme looks playful yet signup system triggers server error 500 unexpectedly
Users exploring online shopping environments often appreciate systems designed to minimize effort while maximizing browsing efficiency and clarity teal cove product studio providing quick loading pages and well structured categories that support easy discovery of items across multiple product groups and sections.
During a review of straightforward vendor house websites, I found browse lemon vendor link – The interface feels clean and organized, and browsing is easy with a natural flow between pages.
Across ecommerce sandbox UI evaluations and vendor system prototypes, testers noticed navigation components containing rain harbor vendor market hall access hub node embedded in page flow, and although the rain concept feels peaceful and balanced, the hall suffers from broken image placeholders which negatively affects usability during testing cycles
mintmeadowgoodsroom – Feels well organized, I didn’t face any issues navigating around.
While checking out various marketplace sites and vendor platforms, I noticed a clean layout page where Bay trade harbor hall portal link – The experience is refreshingly simple, and I really appreciate that it avoids aggressive newsletter interruptions.
velvetbrookartisanboutique.shop – I’d recommend this to anyone who loves handmade goods.
During routine checks of experimental ecommerce layouts and template based shops testers notice that the daisy harbor room environment includes daisy harbor access panel which should open vendor listings but instead pushes users straight back to the main landing page disrupting workflow and navigation logic significantly – this redirect issue makes the vendor room effectively unusable in normal browsing conditions
Digital platforms designed for efficiency often include access points that serve as entryways to structured product databases, allowing users to navigate listings with improved speed and clarity goods room access point – This access point simplifies user interaction by offering a direct path into organized listings and reducing friction during navigation processes
While reviewing intuitive commerce hub designs, I came across visit lemon structured hub – The layout is clean and logical, and browsing through content feels effortless and easy to follow.
Across prototype marketplace systems and UI sandbox environments, analysts encountered embedded navigation blocks containing harbor rain market hall vendor staging console hub within page layout, and although the rain harbor ecosystem is intentionally repetitive, the vendor hall still feels like a copied template which reduces usability during testing sessions
People who appreciate modern artisan branding often browse sites like Harbor Lemon Artisan Supply Outlet where items are presented in a clean structured format – The interface makes browsing feel bright, simple, and easy to navigate throughout the store.
While going through different niche directories and marketplace listings, I came across something that felt clean and intuitive, especially where Pine harbor access link appeared – Good experience overall, and everything seems clear and straightforward here, making browsing feel effortless and smooth.
As I continued exploring online commerce platforms and trade hall listings, I came across something that felt playful in branding but still early in development, particularly Harbor acorn trade hall commerce portal – The acorn concept is enjoyable, but the product range is currently quite restricted.
During interface validation of ecommerce sandboxes, testers discovered that the meadow vendor room link appears embedded within content blocks, and although the meadow concept feels relaxing, SSL certificate warning notifications disrupt user confidence significantly during live preview sessions testing phases
Users engaging with digital trade hubs prefer well structured environments that categorize listings effectively, allowing them to browse smoothly and compare different vendor offerings with minimal effort vendor lounge exploration panel – Vendor lounge feels calm with well structured product categories available, creating an easy to navigate experience where users can focus on discovering relevant vendors without distraction
нейросеть пишет реферат [url=https://nejroset-dlya-referatov-24.ru/]нейросеть пишет реферат[/url] .
During an analysis of craft boutique interface design and usability, I noticed open velvet grove artisan design boutique – There is a minor typo in the description, but overall I’m satisfied with the experience and navigation.
1xbet ?ye ol [url=https://1xbet-67.com/]1xbet-67.com[/url] .
During staging reviews of ecommerce marketplace systems and UI prototype frameworks, analysts encountered a central block featuring meadow vendor solar room market console entry node within layout structure, and despite the solar meadow naming suggesting sustainability, the platform lacks eco indicators which reduces user trust during testing sessions
Users who enjoy clean artisan storefronts often explore sites such as Oak Dock Artisan Flow Outlet where products are displayed in a simple layout – The design ensures browsing feels smooth, easy, and visually organized across categories.
As I continued exploring marketplace-style listings and online resource hubs, I came across something that felt structured and fast, particularly with Isle icicle trade link – The platform is nice, and I appreciate how quickly pages load, making browsing feel effortless and well optimized.
While searching online earlier today, I discovered check this link which seemed to be a nice little site, and I found it useful while browsing earlier today because it was easy to understand and not cluttered with unnecessary elements.
While scanning through marketplace platforms and vendor hall hubs, I noticed something that felt visually gentle and mountain-inspired, especially Alpine cove commerce market hall link – The branding feels like a small alpine shop, giving off a cozy and welcoming atmosphere throughout.
In various QA passes on demo shop systems, analysts identified dawn ridge vendor hall access embedded in central page flow, and although the theme feels cohesive, after the dash – ridge scenery looks calming but footer links are dead and fail to respond under any interaction conditions
электрокранизы [url=https://prokarniz17.ru/]prokarniz17.ru[/url] .
After comparing multiple websites earlier today, I discovered try this page and everything looked neat and quite easy, giving a smooth and user-friendly browsing experience overall.
Users evaluating online vendor ecosystems often prioritize platforms that streamline navigation and present categories clearly for easier comparison across listings and services available fern harbor lounge access – The vendor environment is designed to provide fluid navigation with a clean layout that improves browsing comfort and reduces complexity for visitors
While reviewing staging ecommerce vendor systems and UI marketplace templates, analysts noticed a content block featuring orchard vendor solar market house access console node integrated into layout flow, and despite the solar orchard concept suggesting trust and natural growth like a secure eco marketplace, the checkout page is missing security seals which reduces credibility during usability testing sessions and design evaluations
Users who enjoy artisan styled ecommerce layouts often engage with sites such as Fern Ridge Handmade Artisan Bazaar Hub where products are presented in a clean curated format – The layout ensures browsing feels structured, easy to understand, and visually balanced across all product sections.
wheatmeadowmarketroom.shop – Clean layout and simple navigation, makes exploring content really enjoyable.
While going through different niche listing platforms and curated discovery hubs, I came across something that felt clean and reliable, especially when seeing Ivory harbor marketplace entry included – Looks professional overall, and I might recommend this to others as well since everything is presented in a clear and structured way.
While reviewing digital vendor collective marketplaces, I came across visit oak meadow commerce lane collective – The product photos are really clean, and the descriptions are helpful, making products easier to evaluate.
In staged ecommerce environments and design sandbox experiments developers observe embedded routing components inside central layouts orchard vendor room gateway that appears structured but delivers minimal product exposure often showing only a few listings instead of a full catalog experience – Driftwood styling is consistent yet inventory depth remains disappointingly low
As I explored different online marketplaces and vendor directories, I kept seeing repeating branding themes that felt oddly familiar, particularly Harbor commerce vendor alpine hall portal – The similarity made it feel like I was revisiting the same store repeatedly.
While analyzing online trade catalog structures and experimental vendor marketplace layouts for usability insights and UI reference across different samples Dune Meadow browsing portal the experience remained smooth and pages loaded without delay – Organized interface with consistent responsiveness and simple navigation flow throughout session
While reviewing different online vendor platforms and creative marketplace gallery designs for inspiration and usability insights Pearl Cove browsing gallery portal the navigation felt intuitive and allowed smooth movement between sections without interruptions or confusing transitions. – Everything loaded efficiently and the interface stayed clean which made the browsing session feel relaxed and straightforward
Digital buyers often mention that structured marketplaces allow better comparison of items, particularly when navigating through Floraridge Shopping Access – The interface is considered helpful for simplifying selection and improving overall shopping efficiency while reducing time spent on irrelevant items
Across ecommerce UI prototype reviews, testers noted a well executed sun inspired design system with clear brightness and hierarchy, but missing depth in sections such as a href=”[https://sunharborvendorroom.shop/](https://sunharborvendorroom.shop/)” />sun harbor vendor hub room panel where Sun harbor branding is visually consistent while the vendor room contains no descriptive or contextual information for user interpretation
During my exploration of fast loading web tools, I encountered check hiperfree smooth hub – The platform features a clean interface with fast loading performance and smooth functionality that enhances usability significantly.
Users who enjoy streamlined trading experiences often explore sites such as River Harbor Market Insights Hub where content is displayed in a clean layout – The design creates a practical environment where information is easy to interpret and use.
During my time exploring different online platforms, I noticed go to this link which offered a pleasant experience because everything is organized here, making it easy to understand the layout and content flow naturally.
While scanning through niche directories and curated marketplace platforms, I noticed something that stood out for its simplicity and clarity, especially where Ivory ridge vendor hub appeared – Browsing here feels smooth, with nothing complicated or hard to understand, allowing everything to be explored without confusion.
While testing staged online retail systems and conceptual UI designs, reviewers encountered a mid page insertion containing willow drift commerce gateway inside structured layout – the absence of willow tree visuals makes the interface feel unfinished and somewhat placeholder driven in several key content areas
Users exploring vendor-based platforms often value clean layouts that support efficient navigation and product discovery Canyon Harbor catalog browser this site offers a cohesive experience where browsing feels natural and well-organized throughout extended sessions
While going through online commerce lounge directories and vendor listings, I came across multiple amber-toned layouts, especially Amber ridge vendor lounge commerce page – The design feels modern and warm, but the text contrast makes it a bit visually tiring.
During a relaxed session comparing multiple online marketplace interfaces and vendor gallery concepts for structural inspiration and usability testing across several demo environments Dune Meadow marketplace hub the interface remained stable with quick page transitions and a clearly arranged layout that made browsing straightforward – Overall smooth performance with well structured sections and minimal visual clutter throughout experience
solarorchardartisanemporium.shop – Great for gift shopping, everything arrived in one piece.
While exploring various online tools, I discovered look into this and found its simple layout made browsing feel really smooth, with everything easy to access and clearly organized throughout the site.
Users who prefer cozy artisan ecommerce experiences often explore sites such as Flint Artisan Brook Warm House Hub where products are presented in a clean structured format – The design ensures browsing feels comfortable, calm, and easy to follow across all categories.
While evaluating prototype vendor systems with ocean inspired UI themes, reviewers observed a calming teal color structure that supports visual consistency, but identified limitations in content hierarchy around a href=”https://tealcovemarkethall.shop/
” />teal cove market hall showcase portal where the teal aesthetic feels refined and modern, yet the market hall requires additional category grouping to improve clarity during usability testing and interaction assessment cycles
People who enjoy clean structured marketplaces often engage with sites like Sun Cove Goods Hub District Market where items are presented in a bright and easy to navigate layout – The design creates a pleasant browsing experience that feels efficient, intuitive, and visually clear.
After going through several slow-loading websites, I came across see this resource and appreciated how smooth the experience was overall, since pages loaded quickly without any issues or interruptions during browsing.
While browsing different online vendor platforms, I noticed how smooth navigation and well arranged layouts can greatly improve the overall user experience for visitors Silk Meadow vendor room hub everything felt organized and easy to explore without any confusion
Many consumers engaging with digital marketplaces highlight that streamlined layouts improve decision-making, especially when visiting Cove Marketplace Hall which is commonly seen as intuitive and well organized – users often mention easier access to relevant product categories and improved navigation flow.
While searching for well designed Hawaiian retreat listings with scenic charm, I discovered a detailed accommodation page worth exploring recently < scenic kona lodging guide – It provides a smooth overview of the property, blending clarity and comfort with a very welcoming informational tone overall
calmcovevendorparlor.shop – Really nice platform, easy browsing and smooth user experience today
During usability audits of ecommerce demo stores, testers found a mid layout module dune vendor plaza entry node embedded within structure but readability issues arise due to sandy tone blending making text harder to distinguish – Dune aesthetic is cohesive yet accessibility problems persist in multiple screen environments
While going through different niche directories and curated listing threads, I came across something that felt clean and easy to use, especially where Jewel brook marketplace page appeared – This seems useful overall, and I found the content quite straightforward today, making navigation feel simple and direct.
While comparing vendor galleries, platforms with clear organization and logical navigation tend to perform better overall Canyon Harbor vendor center the browsing process here is smooth and helps users locate relevant listings without unnecessary complexity
While exploring experimental commerce directory systems and online trade gallery platforms for structural analysis and usability insights across various references, I came across Plum Cove goodsroom catalog hub embedded in content – Everything feels clean and easy to read, and I could browse sections comfortably while the layout remained consistent and well structured throughout.
As I explored vendor marketplaces and commerce hubs, I noticed a newly built platform that feels early-stage and underfilled, particularly Cove Aurora commerce room goods link – The design is neat, but the limited product listings per category feel somewhat strange.
sageharborgoodsgallery.shop – Looks clean and minimal, easy to find information without confusion.
People who enjoy artistic online marketplaces often engage with platforms like Cove Teal Vendor Artisan Studio Market where items are displayed in a creative and organized format – The design emphasizes artistic expression and structure, making browsing feel smooth, inspiring, and visually balanced across all product categories.
People who prefer organized online emporiums often engage with sites like Grove Timber Supply Emporium where items are displayed in a structured layout – The interface ensures navigation feels smooth, simple, and easy to manage across categories.
During UX evaluation of sandbox marketplace systems, testers noted a cohesive teal harbor visual structure that improves interface flow, but the vendor hall lacks real content when inspecting a href=”https://tealharborvendorhall.shop/
” />teal harbor vendor hall entry gateway where the design remains polished and consistent, yet the vendor hall continues to rely on Lorem ipsum placeholder text which impacts usability during testing and system review processes
During my time browsing different platforms, I noticed <a href="[https://woodharborvendorroom.shop/](https://woodharborvendorroom.shop/)" / go to this link which seemed like a helpful platform overall, so I might visit again sometime soon because everything was easy to understand.
Many online marketplaces can feel complicated, but this really nice platform keeps browsing easy and user experience smooth today Calm Cove product hub I liked how everything was organized and simple to access
velvetgrovecraftboutique.shop – Little typo in description but overall I’m satisfied.
During an exploration of online vendor platforms, I spent time on vendor discovery page which organizes content in a way that encourages easy scanning and relaxed interaction – The experience feels clear, structured, and comfortably simple to navigate
While performing UX audits on experimental shop templates and modular storefront designs, analysts highlighted embedded navigation segments containing dune meadow market hall entry which appears centrally positioned, yet the naming mismatch between soft meadow and harsh desert imagery creates cognitive dissonance – Meadow name contradicts dunes, leading to unclear branding that reduces coherence in product presentation and overall user experience during evaluation testing
Many online consumers mention that organized vendor platforms reduce browsing frustration, particularly when they open Forest Meadow Listing Gateway Portal and they note that the categories feel logically arranged – this design is often considered beneficial for quicker product discovery and improved clarity when comparing multiple items in one session
As I continued exploring different marketplace-style listings and online discovery threads, I came across something that felt structured and accessible, particularly with Cove marketplace jewel page – The interface is pretty clean, and everything is arranged in a logical way, helping the browsing experience feel comfortable and efficient.
Exploring digital vendor environments often shows how fast loading pages improve user satisfaction and usability overall Solar Meadow listing portal this platform allows users to search easily while keeping performance smooth and responsive at all times
While browsing experimental retail and grocery platforms, I discovered a simple concept website that focuses on clarity and minimal structure hope digital retail hub – The design is effective in its simplicity, making the browsing experience smooth and easy to understand overall
While researching curated vendor-style websites and online shop designs, I discovered an interface that included Honey Meadow artisan market space positioned within a structured layout that emphasizes clarity and user comfort – The browsing experience feels relaxed, coherent, and pleasantly simple to engage with across all sections
Users exploring vendor-style platforms often prefer sites with simple layouts and clean interfaces that make browsing effortless Silver Cove shop access point this one provides a smooth experience where content is easy to find and navigation feels natural and clear
1xbet [url=https://1xbet-71.com/]1xbet[/url] .
During casual research into online vendor systems and digital marketplace layouts for UX inspiration and evaluation across different sample references I came across Teal Harbor showcase entry hub – The browsing experience remains clean and responsive, making it easy to move through categories while maintaining clarity, structure and consistent performance throughout the platform
1xbet g?ncel adres [url=https://1xbet-70.com/]1xbet g?ncel adres[/url] .
1xbet [url=https://1xbet-68.com/]1xbet[/url] .
xbet giri? [url=https://1xbet-69.com/]1xbet-69.com[/url] .
While scanning through online marketplace listings and vendor hubs, I noticed a site with a very appealing autumn theme, especially Autumn meadow commerce vendor hall portal – I really like the seasonal design, and hope they add real customer reviews soon.
People who appreciate efficient digital commerce platforms often browse sites like Harbor Teal Commerce Grid Hub where items are structured in a clean and logical way – The interface makes browsing smooth and easy, with categories that are simple to understand and navigate.
Many online marketplaces feel complicated, but this one offers well organized content and simple layout so users can explore everything quickly River Harbor product hub I liked how smooth and easy navigation felt throughout
While going through various online options, I encountered access this site which had interesting content, and I spent some time checking different sections today while exploring its layout and structure.
People who prefer refined ecommerce experiences often explore sites like Cove Golden Vault Harmony Hub where items are arranged in a minimal structured format – The design ensures browsing feels balanced, polished, and easy to navigate across all categories.
During ecommerce prototype testing sessions, reviewers noted a strong timber trail branding approach that enhances rustic appeal, but navigation issues are severe in components such as a href=”[https://timbertrailmarkethall.shop/](https://timbertrailmarkethall.shop/)” />timber trail vendor hall console node where the UI looks natural and well structured, yet the navigation system is broken which reduces user efficiency during usability testing and interaction evaluation stages
While examining staging storefront builds and experimental checkout systems, developers identified a mid layout module featuring brook trade echo gateway integrated into cart workflow, yet quantity updates do not persist after interaction – Echo brook is a strong brand name, but the cart interface does not update item quantities reliably across sessions and device types
During my casual exploration of experimental vendor platforms and modern online marketplace directories, I came across Moon Cove exploration page – The site felt easy to navigate and thoughtfully structured, and I enjoyed how the content encouraged relaxed browsing across multiple sections without pressure overall.
During a casual browsing session across online resource hubs and marketplace listings, I came across something that felt structured and minimal, particularly references like Orchard mint vendor access – The overall feel here is good, and I like that it’s simple and easy going, which makes navigation feel natural.
Exploring digital vendor spaces often shows how simplicity in design improves overall usability and satisfaction Icicle Brook listing portal this platform supports smooth navigation and allows users to browse items efficiently without confusion
While browsing international beverage brands and winery portfolios, I encountered a visually appealing and information rich wine website worth highlighting icewine collection brand hub – The presentation is detailed and engaging, offering a polished view of the wines and their background story
While researching digital vendor platforms and marketplace structures, I discovered a section featuring Honey Meadow curated market view placed within a visually balanced interface that supports clarity – The browsing experience feels steady, warm, and naturally enjoyable for users exploring content
violetharborretaillane.shop – They responded to my email within an hour, nice.
While analyzing digital vendor marketplace systems and experimental trade hub layouts for structural evaluation and UX research across multiple references, I came across Pebble Pine marketplace structure hub embedded mid-content – I enjoyed browsing listings without confusion since everything felt intuitive, clean, and well organized with smooth navigation throughout the experience.
Some websites feel slow, but this one keeps performance strong with fast loading and neat organization Silver Harbor quick browse page I appreciated how simple and efficient the browsing experience felt throughout
bayharbortradehouse – Almost identical to another bay domain, bit confusing tbh.
Some websites feel messy, but this one keeps smooth experience and clean layout so browsing is very convenient overall today Plum Harbor quick browse page navigation felt simple and clear
During my research into vendor exhibition websites, I came across vendor exhibition space which provides a clean layout emphasizing product clarity and smooth category transitions – The experience feels polished, stable, and pleasantly easy to navigate
Users who appreciate clean digital commerce hubs often browse platforms such as Trail Harbor Commerce Smart Hub where products are displayed in a structured and minimal layout – The design ensures navigation feels easy, intuitive, and well organized for quick browsing and discovery.
During my search for well-designed websites, I stumbled uponclick this design example which felt refreshing, as the design feels modern and neat, definitely stands out nicely with its clean structure and balanced layout throughout the pages.
Users who appreciate structured marketplace design often browse sites such as Granite Vault Orchard Core Hub where products are presented in a simple organized layout – The interface makes browsing feel secure, consistent, and easy to navigate throughout the store.
While performing a structural review, I noticed go here now – the social links in the footer appear interactive but ultimately fail to function, offering no real value to users browsing the page.
Across ecommerce staging environments and UI support modules, developers observed an embedded contact component containing echo harbor vendor support panel integrated into the layout, but despite a polished interface the customer service email system fails repeatedly and generates bounce notifications – Echo harbor branding feels recognizable, however email communication does not reach recipients successfully in testing scenarios
Users exploring vendor directories often prefer platforms that combine structured layouts with clearly defined sections for better usability Violet Harbor shop access point the experience feels intuitive and smooth, making it easy to move through different categories without unnecessary effort or confusion
During a casual browsing session across online directories and marketplace listings, I came across something that felt structured and minimal, particularly references like Cove moon vendor access – The browsing experience feels solid, and I didn’t encounter anything confusing at all, which made everything feel easy to follow.
During my browsing across different vendor-style platforms, I noticed V “browse entry point” inserted in an unusual structure, and the reference to Cicicleislemarketparlor.shop appeared mid-content, while the design still felt modern enough and navigation was quite simple to follow through each section easily.
During casual research into marketplace catalog designs and digital showcase systems I discovered Birch Harbor online vendor hub placed within informational content – the site performed reliably, with fast loading speed and a structured layout that helped maintain clarity while navigating.
While browsing curated experimental web platforms, I came across a site that strongly highlights creative structure and non traditional design flow intermusses design concept portal – The content feels experimental and creatively arranged, making the entire experience feel intentionally artistic and visually distinct
pineharbortradeparlor.shop – Nice interface design makes navigation simple fast and quite pleasant
A modern platform enhances usability, making browsing feel very easy today with a smooth interface overall Linen Cove browsing center I found everything simple and intuitive
While exploring online commerce vendor platforms and directories, I found a site that looks modern but strangely empty in updates, especially Autumn commerce cove room vendor hub – The missing blog content gives a strong impression that the site is no longer active.
People who prefer ocean inspired digital stores often explore sites like Harbor Wave Seaside Commerce Outpost where items are arranged in a calm and clean layout – The interface creates a pleasant browsing flow that feels easy, relaxing, and visually balanced throughout the shopping experience.
A modern interface enhances the browsing experience, and this site offers intuitive navigation with well organized content Sky Harbor browsing center I enjoyed how smooth and easy everything felt while navigating the platform
mintorchardretailatelier.shop – Definitely coming back here for the holiday season.
While searching for a platform that values clarity, I discovered <browse this link which made the experience enjoyable, thanks to its organized layout and easy-to-read information.
People who enjoy modern marketplace structures often explore sites like Orchard Lantern Product Flow Lane Hub where items are arranged in a structured format – The design ensures navigation feels intuitive, simple, and easy to follow across all sections.
During a structural audit of similar platforms, I found follow this link – the duplication in layout and design gives off a strong sense of familiarity that reduces the uniqueness of the experience.
While analyzing ecommerce sandbox systems and nature inspired storefront layouts, testers encountered embedded navigation with elm goods room harbor link within page structure, but product images do not render correctly causing broken visual presentation across multiple listings – Elm trees are strong and steady, yet the goods room suffers from missing image resources during browsing
Online platforms that emphasize both visual appeal and structure tend to provide better browsing experiences overall Forest Meadow vendor navigator this one ensures that users can find important information easily while enjoying a clean and organized layout
As I continued exploring various online resource hubs and marketplace listings, I came across something that felt organized and easy to use, particularly with Harbor moss vendor entry – The site seems decent, and I’ll probably check it again soon since navigation is intuitive and clean.
sageharborgoodsgallery.shop – Looks clean and minimal, easy to find information without confusion.
While reviewing curated trade gallery platforms and marketplace catalog systems for usability testing and structural insights across sample interfaces, I discovered Plum Cove goodsroom browsing portal embedded mid-content – The content remains clear and readable, and I could navigate easily through sections without distractions or unnecessary complexity affecting the experience.
zencovegoodsgallery.shop – Simple interface clear structure makes finding information very quick indeed
A clean platform enhances usability, making exploring products easy and visually pleasant overall Autumn Cove browsing center I found everything simple and clear
While browsing curated personal profile websites, I encountered a polished portfolio page that emphasizes clarity and professional presentation online career portfolio hub – The navigation feels seamless, and the information is clearly structured for easy understanding and quick access throughout
coralharbortradegallery.shop – Great browsing experience overall everything feels organized and easy today
People who prefer handmade ecommerce design often explore sites like Cove Wind Artisan Creative Exchange Hub where items are presented in a neat organized layout – The interface makes browsing feel smooth, visually balanced, and easy to navigate across all product categories.
As I explored various trade hubs and online vendor listings, I came across a berry-themed website featuring Cove berry commerce market house link – It looks fresh and inviting, yet the missing contact page makes it feel incomplete.
While searching for straightforward and organized platforms, I came across explore this site which provided a well-structured experience, allowing users to quickly locate information and move through pages without difficulty.
People who appreciate structured online goods platforms often browse sites like Meadow Juniper Goods Network Hub where items are presented in a clean layout – The design ensures browsing feels accessible, organized, and easy to navigate across all sections.
Clear organization improves usability, and this platform provides structured pages that make browsing smooth and comfortable Sky Harbor vendor navigator I found navigation easy and everything well arranged
As I explored the structure of the platform, I noticed see this page – the branding implies a polished environment, but the actual lounge area is completely empty and visually unengaging.
Users browsing vendor listings typically value platforms where everything loads quickly and feels easy to navigate Sea Cove goods directory the browsing process here is pleasant and helps users access information without delays or confusion
Good usability depends on design, and this platform provides nice visuals with layout that ensures smooth and pleasant browsing Berry Cove vendor explorer I found everything clear and easy to access throughout
In the process of reviewing multiple platforms, I encountered review this link which confirmed everything looks well structured, helping users move around without issues and making navigation very smooth and easy.
Одноразовые медицинские инструменты исключают риск перекрестного инфицирования и экономят ресурсы клиники на дезинфекцию. Весь ассортимент стерильных расходных материалов доступен на https://invasive.ru/.
During a relaxed session analyzing digital marketplace systems and vendor gallery interfaces for structural evaluation and UX comparison, I discovered Sun Cove vendor showcase portal placed mid-content – The interface felt modern and responsive, and I enjoyed how visually pleasant everything looked while browsing through sections without confusion or delay.
People who prefer uncluttered online outlet stores often engage with sites like Stone Harbor Outlet Flow Market where items are displayed in a minimal and organized format – The design emphasizes ease of use and clarity, making browsing straightforward, efficient, and enjoyable for everyday users.
1xbet yeni giri? [url=https://1xbet-giris-69.com/]1xbet yeni giri?[/url] .
While browsing international themed websites with cultural depth, I came across a platform that creatively blends multiple influences in one space cultural fusion concept page – The site feels engaging and diverse, offering a mix of themes that creates an interesting and visually appealing experience overall
During exploration of curated e-commerce gallery designs and storefront interfaces, I noticed Upland Cove artisan market space embedded within a structured layout that prioritizes readability – The experience feels calm, simple, and naturally easy to navigate through content without confusion
While exploring a range of online platforms to compare their structure and presentation, I came across something interesting in the middle like take a look here which felt thoughtfully designed, showing clear effort in its layout and giving a polished impression overall that made browsing quite pleasant.
Users who appreciate green themed ecommerce platforms often browse sites such as Forest Cove Artisan Leaf Outlet Hub where products are presented in a minimal natural design – The layout ensures browsing feels calm, intuitive, and visually appealing throughout the shopping experience.
While wrapping up my session across commerce platforms and vendor hubs, I came across a simple listing site featuring Berry room harbor vendor portal link – It served as my last stop, and I’d describe it as decent but not special.
1xbet giris [url=https://1xbet-giris-68.com/]1xbet giris[/url] .
xbet giri? [url=https://1xbet-giris-67.com/]1xbet-giris-67.com[/url] .
During a structural review, I noticed browse this trade link – the page lacks activity and content, making the trade house seem like a deserted environment rather than a lively marketplace.
A clean interface improves usability, and this platform makes it easy to go through different sections quickly and smoothly Snow Cove marketplace link I found everything clearly organized and easy to access
This platform shows how a simple interface works well, navigation is quick and intuitive today across all content areas Oak Cove market hub everything felt organized clearly
rainharborvendorparlor.shop – Good organization easy navigation helps users find things efficiently today
When reviewing online galleries, structured content is often a key factor in determining ease of use for visitors Elmwood goods browsing center this platform provides a clean layout that supports smooth exploration of sections and content overall
sageharborgoodsgallery.shop – Looks clean and minimal, easy to find information without confusion.
During analysis of vendor showcase platforms and online marketplace lounge systems for usability testing and design inspiration across various references I came across Upland Cove commerce lounge navigator placed within structured content and observed the layout is consistent and visually clean supporting easy browsing and quick understanding – The interface feels modern and neatly arranged, enhancing overall user experience
People who appreciate crisp ecommerce aesthetics often browse platforms like Isle Icicle Cool Breeze Market where products are displayed in a minimal and refreshing layout – The interface ensures usability and visual clarity, making browsing feel calm, smooth, and easy to follow throughout the store experience.
ии для студентов [url=https://nejroset-dlya-referatov-27.ru/]nejroset-dlya-referatov-27.ru[/url] .
People who prefer organized minimal marketplaces often explore sites like Harbor Acorn Outpost Utility Hub where items are grouped in a structured format – The design ensures browsing feels easy, intuitive, and clean while maintaining a simple user experience.
During my time exploring multiple options online, I noticed check this resource and appreciated its structured layout, which made browsing feel effortless while still providing clear and useful information.
While exploring thematic cultural websites, I discovered a platform that integrates global influences into a unified and engaging design structure urban global fusion page – The experience feels diverse and interesting, with a visually appealing combination of cultural elements throughout the site
While analyzing modern digital storefront concepts for inspiration, I found a section containing harbor vendor showcase hub integrated within a structured layout that emphasizes readability and spacing – The experience feels calm and organized, allowing users to explore content comfortably without distraction or visual overload across multiple sections
xbet [url=https://1xbet-giris-70.com/]xbet[/url] .
A well structured interface with clean design greatly improves usability, and this platform makes usage very comfortable overall Acorn Harbor marketplace link I found everything easy to understand and well organized
Users prefer platforms that load quickly and feel smooth, and this site offers exactly that kind of experience Snow Harbor goods directory I liked how responsive everything felt during browsing
During a search for efficient vendor directories, I noticed platforms that emphasize clarity and speed for better user experience >vendor parlor Aurora Harbor the browsing process feels fluid and responsive, making it easier to explore different listings without encountering navigation issues or delays online
While wrapping up a casual survey of online vendor parlor concepts and experimental marketplace designs, I discovered Moon Harbor online parlor entry – The overall browsing experience felt stable and uncomplicated, and I could explore content comfortably without encountering any confusing elements.
Online platforms with intuitive navigation often lead to better user retention and increased engagement levels overall performance gain Valecove Goods Room browsing portal – The interface layout supports efficient movement between sections, ensuring users can locate desired content quickly and comfortably based on user experience evaluations feedback insights data analysis
People who appreciate functional online stores often browse platforms like Stone Outpost Glade Commerce Hub where product presentation is kept minimal and efficient – The design prioritizes clarity and ease of use, helping users quickly find what they need while enjoying a smooth and distraction free browsing experience.
People who appreciate practical digital trade hubs often browse platforms like Harbor Trade Jasper Market Hub where items are presented in a neat organized format – The design ensures browsing feels logical, smooth, and easy to understand throughout the store.
Online platforms should be simple, and this one delivers a nice design overall so pages load fast and feel very smooth Ivory Harbor digital storefront I appreciated the smooth experience from start to finish
After comparing multiple platforms that lacked clarity, I discovered look into this which offered a decent layout and a smooth interface, making it easy to browse without feeling overwhelmed.
Users browsing vendor-style platforms often prefer sites with simple design and fast loading pages that offer a very reliable and easy interface Glass Harbor shop access point this one delivers a smooth and consistent experience overall
cloverharborvendorparlor.shop – Simple layout works well making browsing easy and intuitive today
1xbet spor bahislerinin adresi [url=https://1xbet-giris-71.com/]1xbet spor bahislerinin adresi[/url] .
Exploring vendor directories reveals that responsiveness and clean organization are essential for good usability Raven Grove vendor center this platform maintains a neat layout where browsing feels fluid and interactions are easy to manage
Many shoppers appreciate digital marketplaces that maintain fast responsiveness across all pages, ensuring that browsing remains uninterrupted and efficient, especially when interacting with Vendor platform gateway page system – Everything loaded quickly and consistently, allowing me to browse comfortably without delays or confusion during the entire session.
сео продвижение сайта заказать в москве [url=https://sovross.ru/advertisment/znachenie-kompleksnogo-seo-audita/]сео продвижение сайта заказать в москве[/url]
sageharborgoodsgallery.shop – Looks clean and minimal, easy to find information without confusion.
Many online marketplaces feel cluttered, but this platform maintains good structure so users can browse without confusion or delay Solar Orchard product hub I liked how everything was easy to access and clearly organized throughout
Users who prefer rustic ecommerce environments often explore sites such as Timber Outpost Trail Commerce Hub where products are presented in a clean wood themed structure – The interface ensures browsing feels easy, calm, and well organized with a focus on natural visual flow and simple navigation.
dawnridgegoodsgallery.shop – Clean layout and structure easy to browse different sections quickly
Users who appreciate efficient ecommerce browsing often browse sites such as Glade Ridge Product Exchange Hub where products are presented in a structured format – The interface ensures browsing feels fast, organized, and easy to explore across categories.
In the process of reviewing multiple online sources, I encountered explore this link which provided content that felt both useful and up to date, making it easy for me to browse through different sections without confusion or difficulty.
While researching curated online commerce platforms and storefront designs, I found a structured interface including Pine Harbor shop directory view placed within a clean layout that emphasizes clarity and visual hierarchy – The experience feels smooth, efficient, and easy to follow, allowing users to browse without distraction or unnecessary complexity
After spending time researching niche artisan platforms and curated goods marketplaces that focus on unique handmade items, I found Coastal Meadow Goods Line – The platform offered a very user friendly experience with clear navigation and well structured categories that made it simple to explore a wide range of items while also enjoying a visually appealing layout throughout the site.
Users often look for platforms that keep navigation intuitive while maintaining a clear and organized structure throughout Raven Summit quick browse page this one ensures a smooth flow that helps users explore content efficiently without confusion
Users often mention that modern shopping websites benefit from calm color schemes and well spaced sections that make browsing easier across categories when exploring different products online Velvet Grove vendor hall homepage overall experience felt smooth with pleasant visuals guiding attention naturally through each section without confusion during navigation
People who value modern curated marketplaces often engage with platforms like Stone Golden Collective Luxury Hub where items are displayed with refined branding and thoughtful arrangement – The interface ensures a seamless shopping experience that feels elegant, structured, and visually consistent across the entire collection.
After exploring different boutique-style websites, I found a platform that seemed focused on curated collections and a smooth browsing experience canyon curated store – Product listings were clear and detailed, and navigation felt intuitive, making the entire experience pleasant and efficient overall.
In various sections of the platform visitors come across Cove Marketplace Directory integrated into descriptive text where it helps structure information and improves overall clarity when browsing multiple categories – everything appears well arranged and easy to understand
This type of marketplace works best when good structure throughout the site makes browsing feel easy and well organized like here Jasper Harbor browsing hub navigation felt natural
This type of marketplace platform benefits greatly from organization, and this one makes exploration very convenient for users Garnet Harbor browsing hub I was able to search and browse content easily without any confusion or delays
linencovevendorparlor.shop – Modern design smooth interface makes browsing feel very easy today
Users who appreciate refined ecommerce presentation often browse sites such as Garnet Vault Harbor Showcase Hub where products are displayed in an elegant structured layout – The design ensures browsing feels smooth, consistent, and visually appealing across all categories.
driftwillowmarketparlor.shop – Pretty straightforward design, makes navigation simple for new visitors too.
I wanted some stylish stationery for my office and came upon ElegantPaperStudio – the layout is user-friendly, and browsing through the different options made the decision-making process simple and pleasant.
uplandcovevendorparlor.shop – Modern interface feels smooth with well organized sections throughout site
Online platforms that focus on intuitive design and structured content typically provide a better user experience overall Raven Summit browsing hub this one allows users to navigate comfortably while maintaining clarity and organization throughout sessions
Users who enjoy rustic and handcrafted online stores often browse platforms such as Harbor Trail Artisan Cottage House where items are arranged in a warm and inviting design – The layout ensures browsing feels smooth, friendly, and visually appealing across all product categories.
Many people browsing online stores appreciate when content is arranged in a structured way that reduces confusion and helps them locate desired sections quickly without unnecessary searching effort vendor hall discovery page the interface delivered a smooth experience with well organized elements that supported easy exploration across all visible categories without interruptions or visual overload
Clear design makes this good platform easy, with intuitive navigation and everything feeling well organized and simple Raven Summit vendor navigator I found it very easy to use
In the middle of exploring online platforms, I discovered browse this link and had a good browsing session there, with a layout that feels quite user friendly and a smooth, easy browsing experience throughout.
During late night browsing of online artisan marketplaces I analyzed several platforms for structure and navigation ease and came across Meadow Silk Goods Exchange – Really like the site design it makes browsing enjoyable every time because the pages load quickly and the interface supports effortless movement between categories
Users who frequently browse online vendor sites often look for platforms that feel calm, simple, and easy to navigate Autumn Meadow catalog entry this site provides a relaxed browsing flow where everything is clear and easy to find without effort
Users exploring digital marketplaces often report a pleasant experience using site, everything appears clean and very responsive throughout their browsing session River Cove shop access point everything felt organized and fast
In exploring curated online marketplaces that emphasize handcrafted goods and independent vendor creations, vendor ruby meadow deals – I noticed the site provided a smooth navigation experience, clear product visuals, and helpful customer service that responded promptly when needed during browsing sessions.
Exploring digital marketplaces highlights how important intuitive layouts are for a smooth browsing experience overall River Harbor market access the structure here allows users to quickly locate content and move between sections without difficulty or frustration
Users who prefer clean outlet ecommerce layouts often engage with sites such as Harbor Pine Value Outlet Hub where product listings are grouped for easy understanding – The structure focuses on practical navigation and category clarity, allowing users to browse efficiently while maintaining a well organized and user friendly shopping experience.
In the process of reviewing online commerce directories and gallery-style marketplaces, I discovered Calm Harbor vendor lounge embedded within a visually balanced structure that supports easy scanning – The site feels smooth and calming, making extended browsing sessions comfortable and visually enjoyable without clutter or confusion.
copperharborvendorparlor.shop – Nice structure easy access content is clear and useful today
Users often note that well organized e-commerce platforms create a more enjoyable experience by simplifying navigation and ensuring that all content is easy to access without unnecessary effort Velvet Grove content portal everything appeared clean and structured, making it comfortable to browse through different sections while maintaining a steady and visually pleasing flow
As I explored different online retail sites, I noticed a well-structured shop – everything feels clean and properly arranged, creating a smooth and easy browsing experience overall.
During exploration of the website structure users will come across Digital Market Showcase integrated within descriptive content which supports smoother transitions between different categories while maintaining clarity throughout the browsing journey – the overall setup feels efficient and improves the speed at which users can locate relevant sections or featured items
While exploring multiple online shops for gifts and decor items I came across Lantern Vendor Support Meadow and customer service was helpful I got all my questions answered clearly which improved confidence while shopping and made the entire experience feel smooth and reliable
Exploring digital vendor sites becomes more enjoyable when smooth performance and neat design ensure everything works quite well Stone Harbor vendor center the navigation felt reliable and pages responded quickly during use
CaramelCoveVendorAtelier – Smooth browsing experience, everything feels clean and very well organized.
plumharborvendorparlor.shop – Smooth experience clean layout makes browsing very convenient overall today
Users who appreciate well designed shopping platforms often browse sites such as Upland Commerce Harbor Navigation Hub where products are grouped clearly for easy access – The design ensures browsing feels efficient, structured, and quick across all categories of the ecommerce system.
While comparing vendor platforms, those with helpful organization and clean layouts often stand out the most Rose Cove shop gateway this one delivers a smooth browsing experience that feels intuitive and easy to use overall
floraridgevendorparlor.shop – Nice structured pages provide clear information and smooth navigation flow
I was exploring unique handbags and stumbled on BagBoutiqueOnline – the layout is organized, and choosing fashionable, high-quality bags felt smooth and enjoyable.
Users interacting with online retail platforms value clarity in design and responsiveness across multiple sections and categories PureValue shop access reports indicate smooth performance and well organized navigation structure – The platform inspired creativity and made browsing feel effortless and engaging
I recently explored multiple eCommerce platforms and found this clean retail platform – everything is well arranged, and browsing feels smooth, making it very easy to navigate through products.
Users appreciate how the website balances simplicity and detail, providing enough information while still maintaining a clean interface overall Meadow Harbor Vendor Portal View – this balance helps reduce overwhelm and allows visitors to focus on exploring content at their own comfortable pace.
While reviewing several online marketplaces for general merchandise and everyday essentials I discovered EmberStone Goods Exchange – the checkout process was quick and uncomplicated while product variety covered many categories and shipping updates were consistent making the overall experience feel reliable and efficient
Digital marketplaces benefit from modern layouts, making navigation simple and content clear and helpful overall Bright Harbor item portal I enjoyed how easy everything looked
While reviewing various retail websites, I noticed this accessible store layout – the structure allows users to quickly find and compare items without hassle.
As I explored several online stores, I noticed a well-structured retail hub – the design is clean and intuitive, making product discovery and viewing fast and very convenient for users.
Some websites feel complex, but this one keeps great usability and simple design so everything works perfectly fine indeed here Crystal Harbor quick browse page navigation felt easy and natural
While comparing different vendor showcase environments, I found a system designed around Frost Ridge Merchant Studio that delivers a well-balanced interface with simple navigation and clean visual presentation – it supports quick browsing and makes product exploration feel natural and uninterrupted across all sections.
People who appreciate minimal curated ecommerce platforms often browse sites like Ridge Ivory Vault Style Market where products are arranged with elegant structure and clarity – The interface creates a smooth browsing flow that feels organized, curated, and visually balanced from start to finish.
While reviewing various vendor platforms, those with clear structure and fast responsiveness tend to provide better usability overall Rose Harbor listings page this one ensures smooth navigation across sections and quick access to information without delays or confusion
Many shoppers prefer tools that help them quickly filter and locate products within large vendor databases Wood Cove retail finder tool this improves efficiency by reducing time spent searching and helps users focus on relevant product selections more effectively during typical browsing workflows online today
Shoppers browsing dedicated corners of curated listings and niche vendor collections often find platforms like Violet Listing Corner – where product information is arranged neatly, allowing for easier discovery and more efficient browsing of available items without unnecessary complexity.
floraridgevendorparlor.shop – Nice structured pages provide clear information and smooth navigation flow
While checking several online marketplaces, I discovered this clean product interface – everything is clearly arranged, and browsing feels smooth, simple, and very easy to navigate overall.
People shopping online often seek platforms that provide a seamless browsing experience with quick access to products and simple navigation tools, and this is demonstrated in FastFlow Atelier – the interface is optimized for speed and usability, allowing users to enjoy a smooth journey from product discovery to checkout.
sageharborgoodsgallery.shop – Looks clean and minimal, easy to find information without confusion.
While exploring niche ecommerce stores during a late night search for home decor inspiration I reviewed several websites focusing on clarity and product variety and then encountered Glass Meadow Market House – Everything from browsing to checkout felt seamless, with accurate product listings and a generally smooth user interface that made decision making easy and stress free.
The platform provides a clean interface and easy navigation making browsing pleasant and very smooth today Silver Harbor market hub I found everything well structured
As I reviewed several online shopping sites, I noticed this fast-loading store – it provides a strong selection, smooth performance, and a very professional browsing experience overall.
Users who enjoy warm and cozy ecommerce design often engage with sites such as Honey Cove Soft Vault Studio where products are displayed in a gentle and inviting structure – The design enhances usability and comfort, making browsing feel smooth, pleasant, and visually consistent across categories.
In reviewing vendor-focused digital storefront solutions, I found a structured layout system built around Arctic Vendor Workshop that keeps the interface organized and easy to understand while supporting fast transitions between sections – overall it creates a comfortable browsing environment where users can efficiently explore available content.
I recently explored multiple online marketplaces and found this clean retail interface – it is simple to use, and everything feels well organized for easy browsing.
Exploring digital vendor environments often shows how usability improves overall satisfaction and browsing efficiency Ruby Orchard listing portal this platform allows users to navigate content smoothly while keeping everything simple and accessible
While exploring digital vendor gallery systems and commerce platform designs for usability analysis, I found Kettle Crest marketplace overview page within structured content – The browsing experience felt simple and intuitive, and I could easily access everything without confusion as performance remained fast and stable.
While reviewing various modern marketplace designs focused on usability and aesthetic structure, I came across a cleanly organized layout that featured Linen Meadow gallery portal embedded naturally within a balanced interface that emphasizes spacing and readability – The overall browsing experience feels elegant, fluid, and very easy to navigate, allowing users to move smoothly through sections without confusion or visual strain
Many users who prefer structured online shopping environments often note that clarity and visual organization greatly improve their experience particularly when they find platforms like Maple Crest e-commerce gallery – where browsing feels effortless and product discovery is made simple and enjoyable for everyone.
During my review of marketplace websites, this interesting platform overall stood out, and I enjoyed browsing different sections today here with ease Jewel Brook browsing portal navigation felt simple and intuitive throughout
As I continued browsing different shopping websites, I discovered this clean vendor hub – everything loads quickly, product presentation looks good, and the overall layout feels organized and simple.
While exploring different market gallery websites and digital storefront access points, I came across a platform where Kettle market gallery access – seemed fairly reliable and possibly useful later on. The navigation experience felt consistent, with clear separation between sections and minimal visual clutter.
riverharbormarketparlor.shop – Well organized content simple layout easy to explore everything quickly
During a late evening session exploring ecommerce platforms for curated goods I evaluated product clarity and checkout flow and came across Juniper Harbor Goods Studio and items arrived quickly while site navigation is smooth and very intuitive too which made browsing comfortable efficient and pleasant throughout the entire shopping experience without confusion
While browsing through different online marketplaces during my free time, I came across something interesting like this smooth shopping hub – the interface feels clean and modern, making it very easy to navigate and explore products without confusion or delay.
Users who prefer cohesive and refined online stores often explore platforms such as Gilded Stone Collective Market Hall where items are presented with elegant structure and premium visual identity – The design enhances product appeal through clean layout choices that make browsing feel organized, luxurious, and easy to navigate.
While browsing different online resources for local and niche marketplaces, I found that the platform embedded here provides useful categorization and navigation tools, making it easier for users to compare options when the Granite Orchard Hub appears within the listing, especially when exploring curated product selections and vendor profiles that help streamline decision making for shoppers.
When exploring digital vendor spaces, usability and simple structure are key factors in a smooth user experience Ruby Orchard goods portal this platform ensures that navigation remains easy and content is always accessible without complications
During casual browsing of experimental marketplace systems and vendor gallery platforms for usability insights and structural review I discovered Harbor Moss curated trade explorer panel and quickly recognized the layout is well organized and easy to follow with clear hierarchy and responsive elements that make the browsing experience very straightforward for users overall – Clear structured browsing with stable fast performance
Online buyers often appreciate websites that reduce waiting times and provide intuitive navigation for a better overall shopping experience, and this is seen in PrimeCove MarketHub – the system is designed to be responsive and well organized, helping users browse efficiently and complete purchases without unnecessary complications.
Many users browsing curated artisan shops mentioned positive experiences with services like Harbor Vendor Craft Portal – where the diversity of products was notable and shipping speed consistently exceeded expectations, contributing to a generally positive shopping experience.
honeymeadowmarketgallery.shop – Warm visuals and clean layout create pleasant browsing experience overall
Users browsing vendor-style platforms often prefer sites with clean layouts and clear structure that help them browse different sections quickly Dawn Ridge shop access point this one delivers a very efficient and comfortable browsing experience
As I explored several online stores recently, I noticed a simple vendor marketplace – the browsing experience is smooth and intuitive, and items are clearly shown, making everything easy and convenient today.
I recently explored multiple eCommerce platforms and found this clear product site – everything is designed with simplicity in mind, making shopping smooth and intuitive overall.
During my search for simple and organized websites, I stumbled upon click here to view which had a nice overall structure, making everything easy to access and view in a smooth and user-friendly way.
While navigating through several digital marketplaces, I encountered this smooth shopping platform – everything feels well structured, making it easy to browse items without confusion or delay.
During casual exploration of online stores for decorative products I came across Nightfall Vendor Market Gallery and enjoyed browsing their collection everything is organized and clearly labeled today making the entire browsing process feel structured intuitive and very simple to follow from start to finish
Users who prefer elegant ecommerce presentations often explore sites such as Ginger Galleria Stone Showcase where products are arranged in a clean and fluid gallery layout – The interface emphasizes visual engagement and smooth navigation, ensuring browsing feels natural, stylish, and easy to follow across all sections.
Users often prefer platforms with a very clean interface because it allows easy access to information and smooth browsing Cotton Grove catalog entry I found everything easy to locate
While reviewing multiple ecommerce vendor platforms for usability and interface structure I focused on layout consistency navigation clarity and product accessibility across sections MarbleCove Commerce Studio Hub the great layout made everything simple and I enjoyed how easy it was to navigate making browsing smooth and comfortable throughout the experience today
Across multiple platform comparisons, efficiency and layout design stand out, and Harbor Foundry Vendor List – The system performs smoothly with well-structured categories and fast loading times, helping users move through different marketplace areas without delays or confusion.
Many people appreciate platforms that maintain a smooth interface while keeping pages well structured and easy to browse Sage Harbor vendor navigator this one delivers a pleasant experience where users can navigate content effortlessly
While reviewing multiple digital marketplace examples and vendor catalog systems for UI inspiration and structural study, I discovered Olive Harbor trade navigation board embedded mid-content which made browsing feel steady and predictable – The interface is clean and efficient, helping users understand everything quickly without unnecessary effort or confusion during navigation.
glassharbortradegallery.shop – Simple design fast loading easy to use interface very reliable
While comparing different online trade marketplaces and curated product galleries, I needed help understanding a few features and got prompt replies, Snow Harbor Commerce Display Hub which made my overall browsing journey feel structured, helpful, and pleasantly straightforward from start to finish.
While exploring various retail platforms, I found this smooth marketplace interface – everything feels well structured, and shopping is simple, fast, and very user friendly overall.
waveharborvendorparlor.shop – Smooth performance and tidy layout make site very easy today
During casual exploration of online vendor hubs and marketplace listing pages for study purposes, I browsed several categories and noticed Rose Harbor trade display page placed naturally within the text – I enjoyed checking this out, content feels simple and informative, and everything felt structured and logically arranged for easy use.
I recently explored multiple eCommerce platforms and found this stable shopping hub – the performance is fast and reliable, making browsing feel smooth and stress free.
Users who enjoy organized ecommerce environments often explore platforms such as Acorn Harbor Vendor Central Hall where products are grouped in a structured and easy to browse format – The interface focuses on clarity and efficiency, making the shopping experience intuitive and visually consistent throughout the site.
In the process of reviewing e-commerce UI patterns, I interacted with a streamlined system presenting Vendor Foundry Selection Portal as part of its interface – navigation was smooth, items were well organized, and the browsing experience felt clear, fast, and consistent across all sections of the platform.
During casual browsing of ecommerce marketplaces for artisan goods I explored several platforms and came across Harbor Vendor Pearl Market – Shopping experience was excellent, site loads fast and feels reliable which created a positive impression due to fast loading pages and a well structured layout that made browsing simple and efficient
Digital shopping environments are most effective when well structured, and a relevant example is SmartCart Unified View which organizes products in a way that simplifies exploration and allows users to find items quickly – The layout reduces unnecessary steps and improves overall usability significantly.
Users browsing vendor listings typically value platforms where navigation is simple and information is quickly accessible Sea Cove goods directory this site provides a smooth browsing flow that helps users find relevant content without confusion
In various comparisons of digital storefront systems and e-commerce user experience trends, structured design plays an important role, and HarborCove Retail Atelier Space is often referenced – users describe the system as responsive and easy to navigate, with clearly separated sections that support efficient browsing sessions.
During a relaxed browsing session focused on marketplace catalog systems and online vendor platforms for research and inspiration across multiple examples, I encountered Pebble Creek marketplace flow viewer inside structured content – I enjoyed exploring the site because the layout felt intuitive and every section was clearly presented for easy navigation and understanding.
While evaluating several online artisan galleries offering curated goods and boutique style collections, I noticed differences in presentation and browsing structure, Sky Harbor Craft Corner – The color scheme was attractive and navigation was straightforward, allowing me to explore different categories without any difficulty or confusion.
As I reviewed several online shopping sites, I noticed this clean commerce interface – the design is minimal and clear, making product browsing and comparison very straightforward.
birchharborvendorparlor.shop – Simple structure ensures quick access to useful information always here
москва продвижение раскрутка сайта александр [url=https://infosport.ru/partners1/raskrutka-i-prodvizheniya-sayta-sovremennye-metody-seo-smm-ppc-i-email-marketing]москва продвижение раскрутка сайта александр[/url]
продвижение сайта рязанский проспект [url=https://avtosreda.ru/news-pubs/prodvizhenie-v-google-i-yandeks-osnovy-prakticheskie-sovety-i-effektivnye-strategii/]продвижение сайта рязанский проспект[/url]
During my review of boutique-style online craft platforms, I accessed boutique craft explorer and observed a smooth and structured layout that enhances product visibility – it offers a comfortable browsing rhythm that feels both modern and approachable
In my review of online marketplace systems I focused on product visibility interface design and browsing comfort across categories MeadowCove Vendor Market Studio everything felt clean and structured and the easy checkout process keeps items clearly displayed and well organized improving user satisfaction throughout
Users who enjoy efficient ecommerce systems often explore sites such as Chestnut Vendor Harbor Market Hall where product listings are arranged for easy navigation and fast understanding – The layout focuses on structured presentation and smooth browsing flow, making it simple for users to locate items without confusion or unnecessary visual complexity.
While reviewing vendor exhibition sites I came across Quick Ridge vendor exhibition link – pages loaded quickly and the structure felt consistent, making it easy to move through sections during the entire browsing session smoothly.
During a casual search across various eCommerce platforms, I discovered this stylish shopping page – everything is arranged in a visually pleasing way, making it easy and enjoyable to browse through products without confusion or clutter.
Many online users appreciate platforms that combine simple structure with strong readability for better usability Sea Meadow item portal this setup creates a smooth browsing experience where content is easy to locate and understand
While comparing different online shops for home and lifestyle products I noticed a platform that Ginger Cove Selection House was very user friendly with smooth navigation and clearly labeled product sections – Great structure overall browsing felt effortless and very well organized experience too today
While researching experimental commerce platforms and vendor display systems for structural comparison I encountered Bay Harbor Marketplace Overview Page in the middle of supporting content – The interface felt highly responsive, and page loads were quick, maintaining a clean and efficient browsing flow.
While comparing different shopping platforms for interface quality I noticed a smooth and responsive design that made category switching effortless and intuitive CoveBerry Commerce Point and I easily found what I was looking for without any complications during navigation or product exploration.
In many user experience reviews of e-commerce environments, emphasis is often placed on how easily customers can move between categories and locate relevant items without unnecessary friction or overwhelming design elements Stone Collective Browse Portal – the platform is described as smooth and efficient, allowing quick scanning of listings and simple comparison of different products across structured sections.
Looking for DIY craft kits, I discovered CreativeKitsCorner – the site layout is clear, exploring fun projects was smooth, and selecting kits that engaged my children was simple, enjoyable, and satisfying.
As I explored several online stores recently, I noticed a smooth commerce collection – the product selection is nice, and the interface feels structured, intuitive, and easy to browse without confusion.
In the process of reviewing digital marketplace systems, I discovered a section containing Coral Harbor vendor network plaza embedded within a clean interface that emphasizes spacing – The browsing flow feels calm, structured, and very easy to navigate without confusion
During a casual search across multiple eCommerce platforms, I came across this clean shopping interface – everything is well organized and easy to navigate, making it simple to locate items quickly without any confusion during the browsing process.
E-commerce platforms succeed when they offer straightforward browsing experiences that do not overwhelm users, and a relevant example appears within the interface at TrustBuy Selection Hub which structures product listings in a simple and accessible way – The improved organization helps users focus on items that matter most without unnecessary distractions or confusion
Exploring vendor directories highlights how clean interfaces improve usability and overall browsing efficiency significantly Silk Grove vendor explorer this platform keeps everything organized and ensures users can move through sections without confusion or delay
Many reviewers of educational and creative platforms emphasize how important smooth performance and structured layouts are for maintaining user engagement and motivation over time Pure Value Idea Portal – the platform offers a stable and inspiring environment where users can comfortably explore, learn, and develop new ideas through a clean and interactive design.
This kind of marketplace works well because good browsing flow ensures everything is organized clearly and very efficiently throughout Meadow Harbor browsing hub interface felt simple and logical
Shoppers who frequently explore online trade directories often value usability because it helps them locate products faster and compare options easily, especially on Harbor Alpine Trade Room where the checkout process felt smooth and efficient allowing users to complete orders quickly and confidently – The checkout process felt smooth and efficient, allowing users to complete orders quickly and confidently.
In my evaluation of ecommerce systems I studied performance optimization layout clarity and overall user interaction quality BayHarbor Commerce Showcase Loft the platform was fast and responsive and everything looks professional making it easy for users to browse and explore products efficiently
While reviewing different curated online trade platforms and handmade goods marketplaces, I focused on interface responsiveness and product organization, Upland Harbor Market Desk – The browsing experience felt well balanced and user friendly, with simple navigation and a comfortable layout that made exploring items feel easy and stress free.
website should appear in the middle of line not in the start or end
Поиск по номеру телефона [url=https://finderphone.ru/]finderphone.ru[/url] .
In usability testing of online marketplace layouts, I explored a design that felt modern, responsive, and easy to navigate across all sections BB Market Foundry Center – the browsing experience was efficient, with well organized product listings that made it easy to locate items without confusion or unnecessary scrolling.
интернет раскрутка сайта [url=https://progorodsamara.ru/interesnoe/view/kompleksnoe-prodvizhenie-saytov-v-moskve-chem-professionalnoe-seo-luchshe-lyubitel-skogo]интернет раскрутка сайта[/url]
продвижение сайтов коломна [url=https://komionline.ru/news/prodvinut-sajt-v-moskve-kak-provesti-polnoczennyj-audit-pered-prodvizheniem]продвижение сайтов коломна[/url]
While checking several online marketplaces, I discovered this streamlined shopping page – everything works smoothly, and the checkout process is simple and very efficient overall.
linenmeadowmarketgallery.shop – Elegant interface with smooth flow makes navigation very easy overall
аренда катера [url=https://arenda-yakhty-spb-9.ru/]аренда катера[/url]
While exploring various online educational resources and creative marketplaces that aim to improve user engagement and discovery experiences across different topics Pure Value Outlet Hub – the platform stands out as an engaging environment that encourages exploration and idea development with a smooth learning flow and consistently inspiring creative tools for users at all levels.
аренда катера на 5 человек [url=https://arenda-yakhty-spb-8.ru/]arenda-yakhty-spb-8.ru[/url]
As I explored different digital storefronts, I found this smooth vendor hub – the layout is clean and modern, making browsing simple and very easy to navigate overall.
яхта на 3 часа [url=https://arenda-yakhty-spb-10.ru/]яхта на 3 часа[/url]
During a search for pet accessories, I came across PawsAndWhiskersShop – the selection is thoughtful, navigating the categories is smooth, and finding practical yet stylish items for my pets was effortless.
As I continued reviewing different eCommerce options, I briefly visited open and see and enjoyed browsing the store, noticing that the products are both appealing and priced reasonably well.
While browsing through multiple eCommerce options, I came across this efficient shopping page – the layout is clear and simple, helping users view products without confusion or clutter.
While casually browsing through online stores, I discovered visit this easy shop and saw that everything feels smooth and quick, making it very easy to browse through products without any complications.
During evaluation of modern e-commerce interfaces for research purposes, I explored a clean and responsive system that felt very user friendly, including BirchCove Digital Market – The platform shows products in a structured layout with clear categories, making it easy to understand offerings quickly while maintaining a visually balanced and pleasant browsing experience throughout.
In my search for well designed marketplace portals, I found Velvet Brook curated trade corner embedded within a visually clean layout that highlights sections evenly – The experience feels calm and controlled, making it easy for users to move through categories and focus on content at their own pace
While testing ecommerce interfaces I focused on navigation design responsiveness and overall ease of product discovery across systems CalmBrook Vendor Showcase Studio the platform was clean and smooth and I found what I needed without any trouble which made browsing very comfortable and intuitive
During my evaluation of ecommerce layouts I found one platform where VendorCloud Showcase – It provided a very clean browsing experience with fast response times and simple menu organization, allowing me to explore different categories without confusion and making the overall interaction feel efficient, stable, and professionally structured from start to finish.
snowcovegoodsgallery.shop – Cool minimal design helps users browse content without confusion today
users exploring online stores frequently appreciate websites that maintain fast performance and logical layout design helping them navigate product listings without difficulty or delay fast browse system known for its clean and efficient structure – it provides a seamless shopping experience that feels natural and well organized across all categories and sections available
In various discussions about modern digital marketplaces and user interface design, reviewers often emphasize responsive layouts and structured browsing systems that reduce confusion during product searches when interacting with Hazel Harbor Market Studio – Smooth browsing experience, products are clearly displayed and accessible, making it simple for users to locate items quickly and compare different offerings without unnecessary delays or clutter.
PFL heads back to the Sanford Pentagon on May 2, 2026 with a roster that is built around pound-for-pound division placements as well as well-known veterans from the Bellator days.
During a general browsing session, I came across this clean marketplace corner – the layout is simple, and I found products quickly and easily without confusion or stress.
Searching for office décor ideas led me to WorkSpaceWonders – the variety and layout make it effortless to find practical items that still add a touch of personality and charm.
In the middle of checking out multiple shopping websites, I came across this easy navigation store – the interface is simple and clean, making browsing and shopping feel very smooth and intuitive.
In between reviewing multiple shopping platforms, I explored discover this market and found some interesting products that caught my attention, so I might revisit the site again soon.
As I explored different eCommerce websites, I stopped at enter modern shop hub and noticed a modern layout that looks nice, with products displayed clearly and attractively for better user experience.
Hey, if you’re betting the heavily favored fighters just because there’s an Australian flag beside their name, you might as well light your money on fire inside the UFC Perth event instead.
In my recent exploration of ecommerce storefront designs I reviewed a system that stood out for its clarity and responsiveness where Vendor Foundry Exchange – Product browsing felt smooth and efficient with no lag, and the entire experience felt well structured and user centered.
Shoppers who value structured online experiences often highlight platforms that offer clear category separation, such as modern cart view – The site is generally seen as user focused, providing quick access to various product sections while maintaining a simple and organized browsing experience throughout
In reviewing modern vendor hub designs, I came across Clover Harbor online vendor hub integrated into a structured interface that prioritizes clarity and smooth navigation flow – The overall browsing experience feels steady, minimal, and pleasantly easy to use.
While testing different e-commerce UI prototypes, I came across a clean interface that made browsing comfortable and fast with well defined product sections and categories Birch Harbor Guild Market – Loved the variety available, everything is easy to explore and understand, and the overall design made it simple to locate items without unnecessary effort or complicated navigation steps.
https://paragraph.com/@rahilusi115@gmail.com/fade-the-hype-my-best-bets-for-ufc-perth
The UFC’s Perth show is always a massive vibe, however if you want to leave with real winnings, you have to pull apart the “Aussie, Aussie, Aussie” crowd chants from the actual stylistic math.
/>purevalueoutlet – Inspiring and interactive site, perfect for learning and creating new ideas. Generate 20 variations following all rules above.Make sure that each line is 40 words minimum and the website should appear in the middle of line not in the start or end
users comparing ecommerce websites often prefer platforms that offer clean interfaces and fast navigation allowing them to compare products easily and browse categories without confusion or delay edge cart explorer view recognized for simplicity – it provides a seamless browsing experience where users can move through categories easily while comparing items in a clear and structured shopping environment
When reviewing online shopping ecosystems focused on usability enhancements and design consistency across platforms HoneyCove Commerce Clarity Center evaluators report that clear visual organization supports faster navigation – shoppers enjoy a more relaxed and efficient browsing experience overall.
Users browsing vendor platforms often prefer an elegant layout where information is easy to find and read Gilded Cove shop access point this one delivers a clean and organized experience
In my review of online marketplace interfaces I analyzed visual hierarchy product organization and overall user engagement across categories GingerCove Vendor Market Hub the experience felt stable and well structured and shopping feels easy and enjoyable overall today creating a comfortable and seamless browsing flow
UFC Perth on the 2nd of May, 2026 with a lineup topped by high-stakes technical striking combined with a blend of seasoned veterans going up against up-and-coming local fighters.
Many customers exploring curated handmade goods and unique decor pieces often discover this platform while browsing different artisan marketplaces online Velvet Grove Vendor Gallery because it offers a wide selection of creative listings from independent sellers around the region and beyond – Shipping is consistently quick and communication feels clear and professional throughout the entire buying experience.
In the course of evaluating online marketplaces with emphasis on visual structure and usability, I found that grid layouts enhance clarity and improve shopping flow across categories, which became evident when analyzing smart grid layout hub – The interface presents products in an orderly grid format that makes browsing feel simple, intuitive, and well structured overall.
While checking different digital marketplaces, I noticed streamlined vendor platform – pages open quickly and all categories are arranged neatly, allowing users to access items without delay, making the whole experience feel simple, fast, and well organized for everyday browsing needs.
While browsing through multiple online resources, I discovered open this useful link and found it to be fairly reliable, with a clean presentation that makes it worth revisiting for future updates or insights.
During a routine check of online marketplaces, I discovered open this page and noticed how well it balances simplicity with speed, offering a distraction free interface that ensures pages load quickly and navigation remains effortless throughout.
During my exploration of eCommerce websites, I encountered see this axis shop and found an interesting selection of goods, arranged in a clean and neat structure that improves browsing comfort.
During comparison of multiple ecommerce user interfaces focused on accessibility and speed, I tested a site where CloverCove Commerce Bridge Studio – Navigation was intuitive and fast allowing smooth movement between pages and a consistent browsing experience across all sections today.
Online users searching for better deal aggregation platforms often come across websites designed to simplify browsing experience such as shopping catalog site a resource that helps people quickly identify available offers while keeping the interface straightforward and easy to understand for all types of shoppers
While conducting comparative research on ecommerce UX and simple design systems, I observed that basic layouts improve user satisfaction by keeping navigation clear, which was clear when testing easy navigation shopping hub – The design feels clean and structured, allowing users to browse products effortlessly.
In the process of reviewing modern marketplace UX layouts and curated shop platforms, I came across Snow Cove artisan market hub placed within a clean interface that emphasizes visual balance – The experience feels calm, simple, and easy to navigate without unnecessary complexity or confusion
In the process of examining a range of online stores for general usability and design clarity, I reviewed online store general info page – The overall layout seems fairly intuitive, with organized product categories and a browsing flow that helps users locate items efficiently while maintaining a clean presentation style.
During my search for interesting online stores, I came across this organized vendor hub – the experience is very nice, with fast loading pages and efficient system performance overall.
While conducting comparative research on ecommerce platforms emphasizing smooth marketplace flow, I noticed that structured design improves usability significantly, which stood out when testing easy commerce browsing center – The marketplace design feels smooth, and product browsing is simple, quick, and intuitive.
eva casino
Если нужен eva casino telegram, лучше использовать проверенные каналы. Я тестировал разные варианты. Этот оказался стабильным. Использую
During comparative research of e-commerce systems, I interacted with a layout that emphasized performance, clarity, and structured product navigation Bright Trading Foundry Center – Fast loading pages made browsing seamless, and the shopping experience felt stable and reliable, supporting easy discovery of items without technical slowdowns.
People browsing through structured online marketplaces sometimes reach Raven Grove product showcase gateway which sits inside a neatly arranged interface that supports quick exploration and easy understanding – I personally found the navigation simple and exactly what I expected from such a platform
During usability testing of online shopping systems focused on smart buying flows, I discovered that intuitive navigation significantly enhances user satisfaction, especially in large product catalogs, which became evident when exploring easy smart shopping portal – The platform provides smooth navigation that makes browsing simple, clear, and easy to follow for all users.
HoneyCoveVendorStudio – Clean interface, everything loads fast and works very smoothly.
As I explored different digital storefronts, I found this easy navigation marketplace – the selection is diverse, and browsing feels smooth, simple, and very easy for users.
While comparing different platforms that varied in usability, I noticed open this link and found it to be refreshingly simple, with a layout that supports quick navigation and makes accessing important details feel effortless.
While reviewing different ecommerce storefronts during usability testing sessions I noticed a platform that focused heavily on clarity structure and smooth browsing flow across categories Clover Crest Trading Atelier Shop Clover Crest Trading Atelier Shop The product listings are neatly organized with clear labels and categories which makes browsing intuitive and the overall checkout experience feels smooth structured and pleasant for everyday shopping use.
While browsing casually through different stores, I came across browse fast cart hub and noticed the platform feels fast and responsive, making browsing enjoyable and smooth across all product sections.
In between checking multiple websites, I explored discover this site and felt that the collection is decent, with prices that appear fair given the current offerings.
While conducting usability evaluations of ecommerce platforms focused on trust and simplicity, I noticed that reliable hubs enhance engagement and clarity, which stood out when exploring trusted online shopping portal – The experience is smooth and dependable, with simple navigation throughout the site.
While exploring online retail platforms I evaluated design consistency navigation clarity and product visibility across different sections GladeRidge Trading Product Studio the system was intuitive and the collection of items is impressive and everything is neatly arranged and clear making browsing easy and stress free
Many online shoppers prefer websites that guide them naturally through categories, and they often mention product lane finder which is considered easy to use; the structure supports logical browsing paths that make it easier for users to discover items without confusion and improves overall shopping efficiency across the platform.
The platform provides simple design that works nicely ensuring browsing feels quick and easy throughout Cotton Meadow market hub I found navigation very smooth
During evaluation of vendor directory platforms and curated shop systems, I came across Flora Ridge online vendor hall placed within a minimalist layout that enhances usability – The browsing experience feels relaxed, structured, and easy to navigate without overwhelming visual elements.
goodsparkstore.shop – Nice spark in design, shopping feels smooth and pretty intuitive
In the middle of checking out multiple shopping websites, I found this smooth trading hub – everything is neatly arranged, and items are clearly displayed for quick and simple access.
While analyzing ecommerce systems focused on promotional deal sections and price transparency, I observed that structured layouts improve usability and satisfaction, which became clear when testing smart pricing deals center – The deals section feels appealing, and pricing looks well structured, fair, and easy to understand.
While researching online marketplaces for home and lifestyle products I discovered Meadow Jasper Product Hall and spent time exploring its organized catalog which felt easy to navigate – I appreciated how accurate the listings were since they reflected real product details clearly and made shopping decisions much easier overall
People exploring niche craft marketplaces often appreciate when browsing is simple and structured, especially on sites like Cove Vendor Listing Board where items are displayed clearly for quick selection – Buyers often said checkout was smooth and they completed their purchases without encountering any issues.
In comparative analysis of modern e-commerce interfaces designed to improve shopping efficiency through organized layout systems and fast loading architecture Icicle Commerce Trading Grid users consistently note positive experiences – Browsing feels structured and efficient with easy category navigation minimal visual clutter and smooth transitions that help maintain focus while exploring product listings across sections.
During a comparative analysis of online electronics marketplaces, I found a listing called smart tech browsing center – The platform organizes gadget deals clearly, making it easy for users to find interesting and useful tech products without confusion or unnecessary clutter in the browsing experience overall.
People who shop frequently online tend to choose platforms that prioritize ease of use and fast navigation for better results Trusted Grid Cart Shop – It supports a user centered experience designed to make browsing simple while allowing quick access to various product categories
As I explored several online stores recently, I noticed a simple commerce page – the layout is clear and intuitive, making it easy to explore products while enjoying a smooth and pleasant experience overall.
While analyzing ecommerce storefronts I paid attention to responsiveness layout clarity and ease of navigation CoastBrook Commerce Trade Atelier The interface allowed smooth browsing with fast transitions and simple product discovery flow
I had been searching for something straightforward when I encountered access this site, and I found it to be well-structured, allowing me to explore different sections easily while still understanding the content clearly.
During my usual search across eCommerce websites, I stopped at check this trail market shop and as it was my first time visiting, it looks like a decent place to shop online with a straightforward browsing flow.
In the process of evaluating digital shopping environments focused on modern cart design and efficiency, I found that polished interfaces enhance navigation and user satisfaction, which was clear when reviewing modern checkout experience center – The layout looks sleek and organized, offering a seamless shopping experience that feels natural and easy to use.
In the course of evaluating digital storefront usability, I tested a platform that provided a minimal design approach with clear product categorization and fast navigation CoveBright Market Hub – Everything is arranged in a clean layout that makes browsing enjoyable and helps users quickly locate products without unnecessary complexity.
While comparing several shopping websites, I came across open this buying hub and felt that it seems legit overall, with a smooth interface that makes browsing products a stress free and pleasant experience.
People exploring modern shopping platforms often focus on ease of navigation and system responsiveness when evaluating overall usability and performance quality adaptive store link users frequently describe it as a flexible and efficient browsing solution – The platform is known for maintaining fast performance and smooth interaction flow across multiple product categories
During a casual search across multiple eCommerce platforms, I discovered this clean vendor platform – everything is neatly structured, and browsing is smooth, with products organized in a very simple and user-friendly way.
many users appreciate digital stores that emphasize straightforward cart systems making it easier to review selected products and complete checkout without confusion or unnecessary delays simple order cart guide known for clarity – it delivers a smooth shopping experience where users can manage their cart easily and proceed through checkout quickly while maintaining a consistent and user friendly design overall
During analysis of user-friendly online shopping systems focused on simplicity and accessibility, I found that streamlined cart designs enhance usability when working with platforms such as easy purchase basket – The system keeps everything straightforward, allowing shoppers to manage their cart effortlessly and proceed through checkout without unnecessary steps or complications.
Online users interested in curated vendor collections and artisan product listings frequently discover resources such as Walnut Harbor Curated List which aim to present organized product information that allows users to navigate categories with ease and efficiency – The overall experience is described as clean and minimal, making it easier for users to focus on relevant details.
During casual online research into boutique vendor websites, I encountered a marketplace that felt both modern and reliable Caramel Cove Vendor Showcase Gallery and appreciated how each product page loaded quickly, providing enough detail to support informed browsing decisions without overwhelming the viewer experience overall.
In discussions surrounding modern e-commerce design and user interface optimization, clarity and responsiveness are consistently emphasized by usability experts Icicle Atelier Market Portal navigation feels straightforward and efficient, with a layout that supports easy product discovery and ensures users can access relevant categories without confusion or excessive effort.
While reviewing modern online store designs I came across a platform that feels streamlined and visually appealing overall CoastCove Vendor Gallery Space – The layout highlights products in a simple and clean way that enhances user focus and makes the entire shopping experience feel organized, structured, and easy to interact with across all pages.
While comparing online shopping platforms I focused on interface design usability flow and overall browsing experience MarbleCove Vendor Flow Market the system was intuitive and stable and the site feels modern and browsing products is simple and very intuitive improving usability significantly
In the process of evaluating digital marketplaces focused on cost efficiency and value, I found that affordable product systems enhance usability by reducing financial barriers, which became evident when reviewing value savings shopping portal – The prices seem budget friendly and appealing, suggesting a good platform for economical purchases.
While reviewing various retail websites, I noticed this clean commerce page – everything is well laid out, making navigation feel modern, easy, and very comfortable for browsing products.
Online platforms should prioritize clean structure helps users navigate site smoothly and without confusion for better usability Maple Grove digital storefront I enjoyed the overall browsing experience
fernharborvendorparlor.shop – Nice structure and clean design, makes reading content very convenient.
During a comparative study of ecommerce interface design patterns, I found a listing labeled simple gate product center – The platform offers a minimal layout that makes browsing extremely straightforward, enabling users to quickly locate items without confusion or unnecessary visual complexity interfering with the flow.
As I continued exploring various shopping websites, I landed on browse this better shop and saw that the name matches the idea well, making it seem like a good shopping option for general browsing.
In the middle of checking different marketplaces, I paused at take a look here and found that there is a nice variety of items, with navigation designed in a way that feels very user friendly.
In the course of analyzing e-commerce usability patterns, I interacted with a system that provided a clean and responsive interface for product exploration CalmCove Market Studio – The experience is very smooth, and everything is well organized, helping users quickly locate items without confusion or cluttered design elements.
While exploring various online marketplaces during my free time, I came across something like this floral commerce hub – navigation feels very easy, and the shopping process is natural, smooth, and extremely comfortable for everyday browsing.
Some platforms aim to provide the most flexible browsing experience possible by continuously shifting and refining category structures for better usability, particularly when using Ultimate Category Shift Hub which – offers an adaptive shopping environment designed to simplify exploration while maintaining a rich variety of product choices.
users comparing online shopping platforms frequently highlight the importance of simple navigation and decent product variety helping them browse categories without delays or confusion modern plus cart view appreciated for usability – the platform provides a comfortable shopping experience where users can easily discover products and navigate smoothly across all sections with a clean interface design
While conducting usability evaluations of ecommerce platforms focused on cart performance and simplicity, I noticed that direct cart systems improve engagement and clarity, which stood out when reviewing efficient shopping cart center – The cart hub ensures a smooth and fast checkout experience with minimal effort required.
While analyzing ecommerce UX designs centered on open layouts and category clarity, I observed that spacious designs improve navigation efficiency and user satisfaction, which was evident when reviewing open shopping flow hub – The interface looks clean and well spaced, making category browsing feel natural and simple.
I wanted decorative plants and came across BotanicalHavenGallery – the site is user-friendly, the selection is attractive, and choosing pieces that brightened my home was simple, quick, and enjoyable.
As I explored different digital storefronts, I found this efficient shop interface – everything is well structured, loads quickly, and gives a reliable and smooth user experience overall.
Online shopping environments are often judged by how easily users can navigate products and complete purchases without confusion Grid Deals Central Hub – The platform emphasizes quick browsing and structured categories that help users move efficiently from selection to checkout with minimal effort
Across evaluations of modern shopping environments emphasizing structured layouts and usability improvements Icicle Market Isle Depot – Extensive product selection is organized in a user friendly format, making browsing smooth and helping customers quickly identify relevant items.
While researching ecommerce websites for curated décor and lifestyle goods I evaluated multiple platforms and found Stone Harbor Goods Pavilion – Friendly interface items are displayed clearly and easy to purchase quickly making browsing simple fast and very enjoyable with clear navigation and responsive design throughout
In the middle of exploring multiple websites, I discovered visit this platform and noticed how professionally everything was arranged, giving a well-maintained impression that made it easy to browse and access information without confusion or clutter.
While analyzing ecommerce UX systems focused on centralized browsing and category diversity, I noticed that ultra hub structures improve navigation clarity by keeping everything in one place, which stood out when exploring smart ultra product center – The ultra hub feels modern and user friendly, offering many categories in one place that makes browsing smooth and efficient overall.
During a longer browsing session, I encountered check smart cart store and found a clean interface and smart layout that makes it easy to locate what I need.
While navigating through various shopping websites, I came across click to view store and after a short look around I found it quite impressive, appearing to be a decent and user friendly place.
users browsing online shopping deals frequently value platforms that reduce complexity and highlight savings clearly helping them move between categories and find products efficiently during browsing sessions deal smart browse recognized for simplicity and usability – it offers a smooth experience where users can easily engage with attractive offers while continuing to explore more categories without feeling overwhelmed or slowed down by navigation design
While reviewing different digital storefronts, I encountered this organized marketplace site – everything is clearly structured, the design is clean, and browsing is very easy and comfortable.
many users exploring ecommerce platforms often highlight the importance of simple navigation systems that help them quickly move between categories and discover relevant products without wasting time easy park catalog known for its structured layout and clarity – it provides a smooth browsing experience where users can easily locate items and enjoy a well organized shopping journey overall experience
While conducting comparative research on ecommerce UX and structured marketplace design, I observed that organized systems improve navigation and user satisfaction, which was clear when testing fast shopping zone portal – The buying zone is clearly organized, allowing users to locate products easily without confusion.
During evaluation of online vendor platforms, I observed that Sage Harbor Category Explorer Portal sits clearly in the content flow while well organized pages, everything loads fast and feels very intuitive, which helps users move through sections efficiently while maintaining a consistent, smooth, and well structured browsing experience throughout the site.
During a comparative study of online marketplaces emphasizing clean design and usability, I discovered that fresh hub layouts enhance user experience by simplifying navigation, which became clear when reviewing clean interface shopping center – The design looks neat and modern, making browsing feel light, smooth, and easy.
While testing various online retail experiences, I came across a platform that emphasized simplicity and fast navigation through clearly organized categories VendorCalm Commerce Hub – The usability is excellent, and shopping feels smooth, easy, and stress free from the beginning, allowing users to explore products without confusion or unnecessary effort.
Users exploring structured online trade platforms often appreciate when product listings are clean and easy to follow, especially on sites like Harbor Wind Browse Portal where everything is arranged logically – browsing was described as smooth, and descriptions were consistently clear and helpful for decision making.
While exploring various online marketplaces during my free time, I discovered this clean shopping hub – the structure is very well organized, and the shopping process feels simple, smooth, and thoughtfully designed for an easy user experience.
While reviewing several online options, I discovered open this page which provided a decent experience so far, as the content was organized well and easy to follow without unnecessary complexity.
In the process of evaluating ecommerce platforms with user friendly selection systems, I discovered a section called flex product browsing hub – The interface provides a smooth and adaptable shopping experience where users can freely explore options and select products with ease across all categories.
IvoryBrookVendorFoundry – Clean design, shopping feels smooth and very easy today.
As I explored different eCommerce platforms, I stopped at enter base shop and found it to be a simple yet effective store where everything works smoothly without any glitches or delays.
While browsing ecommerce websites for home décor and curated goods I evaluated layout structure and usability and found Orchard Harbor Trade Gallery – Loved browsing here, found exactly what I needed very fast and the clear organization helped me move through product categories quickly while keeping everything simple and efficient overall
During an interface design analysis of ecommerce platforms, I observed a section named simple click shopping hub – The structure is extremely user friendly, allowing fast access to products and reducing friction in navigation so users can enjoy a smooth and stress free browsing experience throughout.
While reviewing various retail websites, I noticed this smooth vendor marketplace – everything loads quickly, and the browsing experience feels fast, efficient, and very comfortable.
услуги seo продвижения низкие цены [url=https://e-news.su/kompyuternye-obzory-i-poleznosti/489441-chto-takoe-prodvizhenie-sajta.html]услуги seo продвижения низкие цены[/url]
users exploring digital retail platforms often prefer websites that offer clear organization and responsive navigation helping them browse large catalogs without unnecessary effort or confusion peak product guide recognized for its user friendly layout and speed – it ensures a comfortable browsing experience where users can quickly discover items and enjoy a well structured shopping environment overall
While reviewing ecommerce platforms that prioritize clarity and simplicity in their layout structure, I noticed that core store designs often improve usability by keeping everything easy to read, which became clear when exploring simple core shopping hub – The layout feels straightforward and minimal, with product listings that are clear, readable, and easy to scan across different categories.
While conducting usability evaluations of ecommerce platforms focused on daily necessities, I noticed that structured stores improve engagement and reduce friction, which stood out when reviewing daily needs shopping portal – The platform is clean and practical, with essential items grouped into clear categories.
As I continued comparing various shopping websites, I came across browse here and appreciated how the items are grouped logically, allowing for a smooth and efficient browsing experience when looking for specific products.
seo продвижение интернет магазина заказать мск seo [url=https://tainaprirody.ru/bez-rubriki/chastnyy-seo-optimizator-samostoyatelnoe-prodvizhenie-ili-net]seo продвижение интернет магазина заказать мск seo[/url]
Consumers often prefer online stores that simplify the purchasing process by offering clear product information and a wide range of categories for better comparison Mega Wide Deals Hub – Wide selection available here ensures shoppers can access reasonably priced items while exploring a well structured marketplace designed to improve convenience and reduce browsing effort
As I reviewed several online shopping sites, I noticed this clean marketplace interface – the experience is simple and smooth, and items are easy to locate quickly today.
While exploring unique kitchen gadgets, I stumbled upon GourmetGadgetHaven – the interface is intuitive, the products are high-quality, and browsing through innovative tools made shopping effortless and enjoyable, leaving me very pleased with my finds.
In the course of evaluating ecommerce user experience designs, I discovered a section called rapid access product index – The platform offers a highly responsive browsing environment where pages load quickly and users can explore products without delays, enhancing overall efficiency and satisfaction during shopping.
Across various marketplace reviews performance and usability are frequently tied to how well platforms structure their product categories and navigation flow Cove Ivory Commerce Link browsing remains straightforward with clean design and responsive layout across pages improving overall usability today
While browsing casually through different stores, I came across browse smart shop hub and found a nice variety of products, which made looking through different options feel easy and pleasant.
While exploring different product websites, I noticed a minimalistic store design – the layout focuses on clarity, allowing visitors to browse effortlessly while appreciating how well each section is presented.
During late night browsing of ecommerce platforms for handcrafted items I explored several sites and found Olive Orchard Craft Gallery and checkout was simple and the product quality exceeded my expectations today which made the shopping process feel smooth comfortable and very easy to understand without unnecessary complexity or confusion
While comparing websites, I found a nice experience overall, browsing content is simple and very pleasant making navigation effortless Pine Harbor listings page browsing felt natural and stable
During a comparative analysis of online marketplaces focused on layout clarity and usability, I discovered that line-based shop designs enhance product visibility and navigation, which stood out when exploring clear path shopping hub – The interface appears structured and organized, making it easy to browse and find items.
many digital buyers value ecommerce websites that improve cart usability and navigation clarity making shopping more efficient and enjoyable across different product categories and sections place easy cart guide widely recognized for usability – it delivers a smooth browsing experience where users can manage cart selections and explore products comfortably while maintaining a clean and organized interface throughout
ForestCoveCommerceAtelier – Clean layout, products are easy to explore and understand.
When evaluating field-style e-commerce layouts and minimal navigation structures, analysts often mention field style retail directory in comparison notes – It is typically described as a directory-like shopping experience where products are organized in a simple structure that allows easy scanning and fast decision-making for users.
While analyzing ecommerce UX designs centered on direct purchasing and usability, I observed that simple checkout flows improve engagement and efficiency, which was evident when reviewing fast purchase navigation portal – The system is easy to use, with straightforward navigation and a clear buying path.
While checking multiple stores for basic necessities, I stopped at open this link and realized it has a practical selection that could be helpful for everyday needs, possibly worth sharing with friends.
While conducting comparative research on ecommerce browsing systems and interface design, I observed that stacked layouts help reduce navigation complexity and improve content visibility, which became clear when testing smart scroll shopping hub – The interface feels clean and structured, allowing users to browse products easily through a stacked and scroll friendly layout.
During a casual search across multiple eCommerce platforms, I discovered this organized shopping site – the layout is very user friendly, and everything is easy to browse without confusion or unnecessary effort.
In the middle of checking various platforms, I paused at tap to open speak store and found products that seem interesting, with descriptions that are clear and easy to understand for quick browsing.
When analyzing modern online shopping experiences, usability and clarity are often considered key factors that influence satisfaction, especially when platforms are designed for quick browsing and comparison, and in this case Ivory Corner Trading Space users benefit from a straightforward interface that keeps navigation smooth and ensures product information is easy to access without unnecessary complexity or confusion.
During usability testing of digital shopping systems focused on cart functionality and checkout speed, I discovered that flexible cart designs enhance efficiency and satisfaction, which became evident when exploring efficient cart checkout hub – The cart is easy to manage, and the checkout flow feels smooth and straightforward.
During casual research of ecommerce platforms for curated home items I compared usability performance and support quality and came across Silk Grove Vendor Gallery – Great selection shipping was quick and customer support was friendly too which made the experience feel intuitive organized and very easy to navigate without any issues
moderndealsstore.shop – Modern design with appealing deals, overall browsing experience feels smooth
In the middle of browsing several online stores, I came across this smooth checkout site – the process of adding items and completing orders feels fluid, with pages responding quickly and reliably throughout the experience.
During a general browsing session, I came across this structured shopping platform – everything is well displayed, the variety is good, and shopping feels simple and very easy overall.
While reviewing ecommerce systems designed for structured browsing and clarity, I observed that shopping hubs improve usability and reduce effort, which was evident when exploring clean shopping flow center – The shopping experience is smooth, and navigating between categories feels simple and well structured.
In some digital marketplace overviews and shopping analysis articles, references such as ecommerce comparison page appear within explanatory sections that break down store features and product listings – It tends to indicate a broad comparison style interface where multiple categories are presented for easy evaluation
москва seo агентство продвижение сайта [url=https://mir76.ru/firms/uslugi/chastnyy-optimizator-saytov-kogda-ego-uslugi-effektivnee-agentskogo-seo/]москва seo агентство продвижение сайта[/url]
продвижение мск сео [url=https://yablor.ru/blogs/tarifi-na-seo-prodvijenie-sayta-kak/8035301]продвижение мск сео[/url]
seo продвижение сайтов в топ москва [url=https://donvesti.ru/news/38092]seo продвижение сайтов в топ москва[/url]
As I continued reviewing different platforms, I briefly visited open and see and even though it was my first time there, the uncluttered interface made a great initial impression.
During a comparative review of online retail interfaces emphasizing clarity and structure, I discovered a module titled organized port marketplace portal – The platform uses a port inspired layout that keeps navigation clean and intuitive, allowing users to browse products easily without confusion or unnecessary complexity in the interface.
When browsing vendor sites, design matters, and this one uses strong layout design for easy navigation and smooth user experience today Harbor Stone goods portal everything felt responsive
While checking several online marketplaces, I discovered this smooth vendor shop – the layout is clean and intuitive, making the user experience very easy and enjoyable.
During my exploration of online marketplaces, I encountered see good trend store and found the trending items look appealing, which makes me consider ordering something from here later on.
Modern online shopping experiences are greatly enhanced by checkout systems that prioritize speed and reduce unnecessary interruptions during payment, and a relevant example is CartFlow Rapid Purchase – It offers a fast and reliable checkout process that helps users complete transactions efficiently while maintaining a smooth overall browsing experience
While analyzing online commerce platforms that prioritize speed, clarity, and user engagement through optimized browsing architecture and system performance, MarketFoundry Brook Explorer is recognized because fast loading pages make shopping feel smooth and dependable, allowing users to navigate effortlessly without interruptions or lag during product exploration.
During usability testing of digital shopping systems focused on performance and quick interaction, I discovered that responsive carts enhance engagement and reduce delays, which became evident when exploring fast response shopping hub – The interface feels highly responsive, allowing users to shop quickly without lag or interruption.
During evaluation of modern online apparel stores, I came across a page labeled fashion trend showcase portal – The interface highlights stylish clothing items in a visually appealing manner, creating a fresh and trendy shopping environment that feels easy to navigate and pleasant for everyday browsing.
While navigating through several digital marketplaces, I discovered a href=”[https://frostbrookvendorfoundry.shop/](https://frostbrookvendorfoundry.shop/)” />this clean commerce portal – the interface is smooth, and shopping feels simple, structured, and very easy overall.
While exploring different online marketplaces for home décor and creative items, I spent a good amount of time navigating categories and product pages and came across Gilded Grove Market Explorer and really enjoyed browsing, everything is organized and easy to understand quickly, making the experience feel smooth and efficient overall for first-time visitors like me.
Many shoppers planning home improvements or restocking essentials often use platforms like home items depot while looking for practical solutions for everyday living – The browsing experience is considered smooth and helps users locate relevant household goods without unnecessary difficulty
During a comparative review of online cart systems and checkout interfaces, I discovered that structured functionality enhances clarity and usability, which became evident when exploring streamlined cart solution hub – The open cart system looks functional, and the shopping steps are clear, simple, and easy to navigate.
In the middle of exploring several digital storefronts, I came across this simple retail platform – the items are displayed in an organized way, helping users navigate through categories quickly and comfortably.
In the course of evaluating online retail platforms focused on usability and structured design, I found that smart hub systems improve shopping efficiency by streamlining navigation, which was evident when analyzing smart retail navigation portal – The platform feels well structured and efficient, allowing users to browse products smoothly through an intuitive and organized interface.
While comparing multiple online stores, I came across open this shopping link and noticed competitive pricing across items, so I am planning to explore more products later today at a more relaxed pace.
While navigating through different eCommerce platforms, I came across click to view goods point and found everything organized well, making category browsing feel convenient and easy to follow.
In discussions about improving digital shopping interfaces, analysts often highlight how responsive design and simplified navigation contribute to better overall performance and user satisfaction across different browsing environments Jasper Cove Interface Studio – The layout is clean and efficient, enabling quick access to products and smooth category transitions.
In the course of evaluating online shopping systems focused on cart optimization and performance, I found that efficient cart solutions enhance clarity and usability, which became evident when analyzing quick cart efficiency hub – The platform feels smooth and fast, making browsing and checkout easy and convenient.
Как правильно указывать NAP-данные при Гео оптимизации сайта? [url=https://geo-optimizaciya-sajta.ru]Гео оптимизация сайта[/url]
[url=https://9online.crforum.ru/viewtopic.php?t=35053]Продвижение сайтов в google[/url] — как связаны CTR в выдаче и позиции сайта?
users comparing ecommerce platforms often highlight the importance of responsive page loading and structured navigation systems that allow them to browse products quickly and efficiently across multiple sections link cart explorer view known for usability – it delivers a seamless browsing experience where users can click through pages effortlessly while maintaining clarity and smooth navigation throughout their shopping session
[url=https://moskovsky.borda.ru/?1-7-0-00013182-000-0-0-1776944928]Разработка сайтов[/url] на WordPress — какие плюсы и минусы для бизнеса?
In the course of evaluating online retail platforms focused on fast promotional access and usability, I found that optimized deal hubs enhance user satisfaction and speed, which was evident when analyzing fast access deal center – The site is quick to load, and the offers feel attractive and helpful for saving time.
While reviewing different digital storefronts, I encountered this clean marketplace interface – browsing is smooth, everything loads quickly, and the design looks very clean and modern overall.
When browsing different e-commerce comparison blogs and analyzing user feedback about retail platforms, shoppers sometimes notice references like deal hunter hub – A straightforward marketplace setup that emphasizes practical discounts and offers a reasonably diverse selection of items suited for everyday purchasing decisions and casual online shopping habits.
While exploring various ecommerce websites for curated products I reviewed performance and layout and discovered Rain Harbor Bazaar Exchange which impressed me with its simple design and smooth navigation that improved browsing speed – Items arrived promptly the site was intuitive and the shopping experience felt efficient clear and very comfortable overall
A well built system ensures good interface design makes browsing straightforward and highly convenient overall for better usability Oak Meadow marketplace link everything was clearly arranged
While exploring ecommerce website structures for design inspiration, I discovered a module titled soft comfort retail hub – The interface is clean and cozy, providing a pleasant browsing experience that helps users move through product listings without confusion or unnecessary design distractions affecting usability.
While browsing casually through apparel websites, I came across browse fashion clothing hub and saw that prices look reasonable, with stylish and modern clothing selections available throughout the site.
During a comparative analysis of online marketplaces focused on simplicity and user experience, I discovered that clean cart systems improve usability and reduce friction, which stood out when exploring simple checkout portal – The design is minimal and user friendly, allowing smooth navigation without any confusion during shopping.
While browsing casually through different stores, I came across browse this marketplace and noticed interesting items that made me curious, encouraging me to return again for further exploration.
People exploring online electronics stores often seek platforms that consistently provide updated listings and trustworthy discount opportunities across categories Smart Gadget Center – Users benefit from frequently refreshed product deals ensuring better affordability while shopping for essential gadgets and modern electronic devices online today
While navigating through different online shops, I encountered this structured marketplace – it offers a clean and professional browsing environment where everything feels well arranged and easy to explore.
Across usability research on online shopping platforms, consistent design structure and predictable navigation are frequently identified as key factors in improving customer experience and retention rates Harbor Trading Showcase Hub – The selection is neatly arranged, allowing users to browse comfortably and understand categories at a glance.
Как [url=https://domashnie-zhivotnye-1.ru]домашние животные[/url] сигнализируют о голоде или жажде?
Online buyers frequently look for platforms that combine simplicity with variety while offering continuous access to affordable deals where smooth discount shopping portal is included in guides – it reflects a streamlined marketplace designed to help users browse efficiently and complete purchases without unnecessary complications or delays in the process.
users comparing ecommerce platforms often highlight the importance of direct navigation systems that reduce browsing time and help them find products quickly without unnecessary complexity quick direct shop explorer appreciated for usability – it delivers a smooth shopping experience where users can move through categories easily and find items without confusion while maintaining a clear and consistent interface overall
Consumers exploring e-commerce websites frequently look for marketplaces that offer structured deal systems and easy browsing where quick shop nest center is included in descriptions and it highlights a platform designed to improve usability while ensuring users can quickly access discounted products and complete purchases without unnecessary delays or complexity.
While exploring ecommerce navigation patterns that prioritize structure and clarity, I observed improved engagement when users interact with platforms such as branch cart explorer – The branching cart exploration system helps users filter products step by step, making the shopping experience more guided and easier to follow.
Many users searching for online savings tools often come across platforms designed for clarity, and an example is online savings center which organizes various offers into accessible sections; overall it is considered user friendly and suitable for those who want a simple way to browse international deals without confusion
While researching different boutique-style ecommerce stores, I found a site that offered a clean interface and structured product listings, and during browsing I noticed Harbor Parlor Coast Market appearing naturally within content sections, and the overall shopping flow felt easy, with quick checkout and a decent range of items worth exploring again later.
In the middle of reviewing shopping websites, I discovered this simple online market and found interesting products available, with browsing feeling fast, efficient, and easy to use without complications.
While conducting usability research on ecommerce navigation systems, I observed a section titled tree flow product index – The platform uses a structured branching layout that simplifies browsing and helps users quickly understand category hierarchy while exploring products in a smooth and organized way overall.
During a comparative analysis of online marketplaces focused on smart product organization and navigation ease, I discovered that intelligent setups enhance user experience and efficiency, which stood out when exploring efficient product marketplace portal – The marketplace structure is smart, with well categorized products that are easy to locate and browse.
While going through online discoveries, I noticed this blunty mill website and after checking it out, it feels quite interesting overall, with a tone that makes it more engaging than I initially expected.
While exploring online marketplaces, I stopped at explore shop rise market and noticed a good first impression, with clean layout and easy navigation that makes browsing enjoyable.
While browsing motorsport charity initiatives online, I came across support racing charity page and interesting concept overall, seems well organized and quite engaging today, presenting a clean layout that connects racing events with meaningful fundraising goals. – The experience feels simple and impactful.
While casually browsing for products online, I discovered visit this page now and saw that the pages open fast, with a decent layout that keeps the shopping experience calm and simple to follow.
In many evaluations of digital commerce systems, researchers highlight that intuitive navigation and structured catalogs reduce user effort and improve satisfaction Brook Market Atelier Flow – Browsing feels easy and very comfortable, with a clean interface that supports smooth exploration of products.
openmarketshop.shop – Open market vibe, lots of items available in one place
Good usability depends on design, and a clean and modern look, everything works well and loads quickly here Opal River vendor explorer navigation felt simple
Consumers exploring e-commerce websites frequently look for marketplaces that offer structured deal systems and easy browsing where quick shop nest center is included in descriptions and it highlights a platform designed to improve usability while ensuring users can quickly access discounted products and complete purchases without unnecessary delays or complexity.
In the middle of exploring various shopping sites, I came across this user-friendly platform – its easy navigation and well-organized product listings create a smooth and pleasant browsing experience.
During an evaluation of budget-friendly online marketplaces, I found that transparent pricing improves shopping confidence when interacting with systems such as smart pricing value hub – The offers appear useful and well structured, giving shoppers a sense of reliability and practical savings opportunities.
In studies of streamlined online shopping experiences researchers often measure how efficiently users locate desired products in catalogs harbor goods navigator referenced in usability reviews – The interface supports fast browsing and clear product categorization for everyday shoppers.
While going through various digital marketplaces, I discovered this simple product store and found it to be a small but practical shop where everything feels reasonably arranged and easy to browse without unnecessary complexity.
During a comparative analysis of online marketplaces focused on promotional pricing and user value, I discovered that deal platforms increase user engagement by showcasing attractive offers, which stood out when exploring smart bargain shopping center – The deals appear very attractive and useful, making it feel like a trustworthy space for users looking to save money while shopping online.
During casual browsing for online gift shops I evaluated product detail accuracy and shipping times before finding Meadow Coral Online Gallery – The items matched descriptions perfectly and arrived quickly which made the whole process feel efficient reliable and something I would gladly repeat in the future
In the course of evaluating online retail platforms focused on cart usability and checkout structure, I found that corner based cart systems help users finalize purchases more efficiently, which was evident when analyzing smart shopping cart center – The design feels practical and well structured, with a checkout flow that is simple, smooth, and easy to navigate overall.
Fashion lovers seeking updated wardrobe inspiration often explore brands that present minimalist aesthetics combined with trendy and versatile clothing options for everyday use Contemporary Fashion Gallery – providing a digital space showcasing stylish clothing collections and modern aesthetic designs that highlight individuality creativity and comfort while aligning with current fashion trends and lifestyle preferences
Shoppers who spend time searching for general e-commerce sites may notice curated lists where this link appears inside descriptive content about stores – The platform seems to present itself as an all-in-one retail space with many product sections and a wide selection designed for different types of buyers.
While exploring web content casually, I noticed this online showcase portfolio and found it while browsing, and the content seems pretty decent, with a clean and straightforward design that keeps things easy to follow.
Shoppers searching for reliable online deals frequently choose platforms that organize discounts into easy to navigate sections for better usability where daily bargain access point is included in guides – it emphasizes a user centered system that makes browsing promotions easier while supporting quick and efficient purchasing decisions for everyday needs.
While browsing general e-commerce pages, I came across straightforward shop platform and the shopping experience here looks simple, clean, and surprisingly intuitive overall, offering users a minimal interface that reduces friction during browsing. – It feels practical, clean, and easy to use.
While reviewing several ecommerce directories for usability and layout design, I came across a labeled section dockmart product gateway – The platform appears well structured, offering organized product divisions that help users quickly identify and explore relevant items while maintaining a consistent browsing experience across pages.
During my search across different online tech shops, I stopped at check this device store and found the products appear interesting, but I still hope the real quality is as good as what the descriptions are suggesting.
While reviewing ecommerce systems designed for structured navigation and step-by-step exploration, I noticed that guided browsing improves clarity and user experience, which stood out when exploring step navigation product hub – The step-based browsing is very clear, and product discovery feels simple and well structured.
Shoppers searching for reliable online marketplaces usually prefer websites that offer verified listings and smooth navigation where affordable trusted deals portal is featured in guides and it highlights a system designed to make online shopping easier while ensuring users can efficiently browse products and enjoy a fast and convenient checkout experience overall.
E-commerce analysts often review shopping cart tools and usability factors when studying how users move from product selection to payment confirmation screens cart usability guide within various testing environments and checkout simulations across platforms – The overall cart experience feels organized, with a focus on reducing friction during final purchase completion.
Shoppers who prefer organized digital stores often appreciate platforms that provide a nice marketplace vibe with clear product variety allowing easy browsing and discovery Harbor Violet Goods Flow Lane Hub – offering a structured e commerce platform where products are organized for clarity and navigation is smooth creating a pleasant shopping experience for users
During analysis of online retail systems focused on global cart usability and product range, I discovered that worldwide cart structures enhance browsing flow and selection clarity, which became clear when testing global cart selection hub – The store follows a global cart layout, providing a wide variety of online products for easy browsing.
As I continued browsing household shopping websites, I discovered this simple essentials platform and liked how everything is easy to find, with a clean layout that supports smooth and straightforward navigation throughout.
While reviewing different digital storefronts, I encountered this smooth browsing platform – everything works in a way that makes locating products quick, clear, and easy for visitors.
In the course of reviewing ecommerce browsing systems, I discovered a section called axis flow product portal – The platform uses a logical arrangement of items that improves navigation and helps users browse efficiently without confusion or unnecessary design distractions across pages.
People exploring modern apparel often seek fashion platforms that combine aesthetic inspired clothing with stylish and versatile wardrobe collections for daily wear Minimalist Style Clothing Hub – providing a curated selection of modern fashion pieces that reflect clean aesthetics contemporary design and wearable comfort designed for individuals who appreciate simple elegant and trend conscious wardrobe options
While browsing through various online stores today, I came across visit kitchen tools shop and noticed the kitchen tools selection looks quite useful, making me consider picking something for home use after further checking different items available there.
During my browsing session across multiple pages, I encountered this simple clone site and after checking it out, it honestly doesn’t look bad at all and might be something worth exploring further depending on user interest.
While reviewing lesser-known concept pages, I encountered this baby bonus informational hub and it seems like a niche idea, but still quite interesting overall, offering a structure that feels simple yet a bit unusual in theme.
This week’s PFL Sioux Falls card at the Sanford Pentagon provides a premium mix of regional standouts and seasoned divisional fighters aiming to secure their standing in the 2026 season.
Many consumers exploring online retail platforms often appreciate websites that highlight deals clearly and reduce browsing complexity where value deal access hub is included in guides and it reflects a structured shopping system designed to help users quickly locate affordable products while enjoying a smooth and intuitive experience across all categories.
many digital shoppers value ecommerce platforms that improve usability through clean design and intuitive navigation allowing them to browse products efficiently without distractions or delays easy zone browse cart widely seen as practical – it provides a smooth shopping experience where users can explore categories easily while maintaining a consistent and simple layout throughout browsing sessions
While casually browsing for online bargains, I discovered visit this discount page and saw that the deals are visually appealing and reasonably priced, which makes me consider buying something soon.
In the course of evaluating online retail platforms focused on modern design and usability, I found that corner cart layouts enhance browsing efficiency and clarity, which was evident when analyzing clean modern shopping hub – The design feels minimal and organized, offering a user friendly shopping experience that is simple and pleasant.
During a comparative review of online value-based marketplaces, I observed that transparent pricing models enhance trust when exploring platforms such as smart value shopping hub – Pricing appears fair overall, and the offers are structured in a way that feels helpful and easy for users to understand.
PFL Sioux Falls is essentially a Logan Storley homecoming celebration disguised as a professional fight card.
people exploring ecommerce options often highlight platforms that balance variety with ease of navigation making shopping more efficient and enjoyable across different product categories plus market view frequently seen as practical and user friendly – it offers a smooth browsing experience that allows users to move through sections easily while maintaining clarity and organization throughout
Individuals who enjoy browsing modern online stores often look for platforms that feature clean interfaces and vault inspired organization to improve shopping flow and usability Cove Vault Product Studio – offering a structured and visually appealing e commerce environment focused on clarity efficient browsing and a creative vault concept that enhances how users interact with product listings online
As I explored different e-commerce pages, I came upon this instant shopping site and found the browsing speed impressive, making the whole experience feel smooth, fast, and very user-friendly from start to finish.
The PFL heads back back to Sanford Pentagon on Saturday for a show featuring as the main event local favorite Logan Storley.
Many consumers browsing online stores often appreciate platforms that highlight multiple product categories and quick checkout systems where smart value cart hub is included in guides and it highlights a system designed to improve usability while ensuring users can quickly find products and complete transactions without unnecessary effort or confusion.
During a comparative evaluation of online shopping platforms focused on navigation flow and usability, I discovered that route-based systems improve clarity and reduce browsing friction, which became evident when reviewing smart route browsing center – The platform guides users through categories naturally, making product discovery simple, smooth, and efficient overall.
People interested in contemporary fashion frequently search for brands that blend minimal design with stylish clothing pieces suitable for versatile everyday wear and creative self expression Aesthetic Clothing Collection – presenting a modern fashion website that showcases curated apparel collections focused on elegance simplicity and trend inspired designs created for individuals who appreciate refined and stylish wardrobe choices
As I continued exploring different online stores, I landed on browse this everything shop and saw it functions as a nice all-in-one store, offering a bit of everything which makes it easy to explore without switching sites.
On Saturday, that home-field dynamic will hit hardest by Florim Zendeli, an athlete tasked with walking into a hornet’s nest to square off against the local favorite, Logan Storley.
While going through different analysis sites, I came across opinion reflection hub and found it quite engaging for a short time, as it presented an interesting perspective that made me think more carefully about the subject matter.
People browsing for convenient online marketplaces often value websites that combine multiple categories into one easy to use interface where everything cart store appears in guides emphasizing accessibility – it supports a smooth shopping journey that allows users to explore different types of products without switching between multiple platforms.
While exploring hotel booking and travel information platforms, I discovered tango themed accommodation page and hotel related content appears helpful, informative, and easy to navigate, offering a smooth browsing experience with clearly organized details about stays and amenities. – It feels simple, modern, and well structured for users.
I recently explored multiple eCommerce platforms and found this consistent retail page – it offers a reliable experience with fast page loads and smooth navigation throughout.
During usability testing of farm oriented online shops, I observed a section marked country harvest field store – The field inspired design organizes content clearly and allows users to browse products in a natural and efficient way overall interface works well.
During a comparative analysis of online marketplaces focused on variety and centralized shopping, I discovered that all-in-one stores enhance user engagement and efficiency, which stood out when exploring diverse product shopping hub – The platform offers a wide variety of products, making it a convenient space for exploring multiple options easily.
During analysis of online shopping platforms emphasizing responsiveness and speed, I discovered that performance optimization enhances usability, which became clear when testing rapid shopping hub portal – The platform is responsive, and pages load fast, providing a smooth and efficient experience.
During my search for mobile accessories, I stopped at check this page and found a decent selection of items, where the designs look modern and attractive enough to catch attention quickly.
As I continued checking digital marketplaces, I encountered this hassle-free shopping site and appreciated how it simplifies the process, making it easier for users to browse and buy without unnecessary complexity.
People who enjoy simple shopping experiences often look for platforms that offer smooth navigation and a diverse range of goods available in a single organized marketplace for better usability Silk Valley Everyday Goods Hub – providing a structured online store where products are grouped together in one place allowing users to browse smoothly and find items quickly across different categories
Online users often seek platforms with clean design and intuitive shopping navigation efficient buying space This makes browsing easier and helps users locate products faster in practical everyday use – System prioritizes user experience with smooth transitions across categories overall experience
Fashion lovers seeking updated wardrobe inspiration often explore brands that present minimalist aesthetics combined with trendy and versatile clothing options for everyday use Contemporary Fashion Gallery – providing a digital space showcasing stylish clothing collections and modern aesthetic designs that highlight individuality creativity and comfort while aligning with current fashion trends and lifestyle preferences
While going through various eCommerce stores, I paused at quick visit pick shop and noticed that selecting items is easy, with a layout that makes browsing feel structured and simple to use.
People searching for practical shopping solutions often prefer platforms that offer essential items with fast navigation where affordable essentials quick hub is featured in descriptions and it highlights a system designed to streamline browsing while ensuring users can easily find products and enjoy a seamless and efficient shopping experience overall.
Consumers looking for efficient online marketplaces tend to prefer websites that offer direct browsing of global goods with minimal steps and fast loading product pages fast track global store helping them discover and purchase items quickly while maintaining a smooth shopping journey – This reflects the growing preference for speed optimized digital retail platforms in modern commerce.
As I reviewed different informational platforms, I discovered this clean email overview site and liked how simple everything is arranged, making it easy to navigate and understand without any confusion.
During my browsing of bakery inspiration pages, I discovered this dessert menu inspiration site and everything looks delicious here, making the site feel very appealing, presenting treats in a way that feels both elegant and appetizing.
While conducting comparative research on online discount hubs, I came across a section labeled discount discovery marketplace view – The interface highlights value driven products, making it simple for users to browse further and discover deals that feel attractive and relevant to their shopping needs.
In the course of evaluating online shopping systems focused on structured layouts and user experience, I found that stacked designs improve browsing flow by presenting items in a logical order, which became evident when analyzing intuitive stacked shopping portal – The layout is clear and well organized, making browsing smooth and easy for users.
users browsing online marketplaces frequently prefer systems that offer guided navigation paths helping them explore products step by step while maintaining a clean and intuitive interface across categories easy trail cart navigator known for usability – it delivers a comfortable shopping experience where users can easily follow browsing paths and move between sections while maintaining clarity and consistent design throughout the platform
While analyzing ecommerce systems designed for guided product exploration and step-based browsing, I noticed that structured navigation improves usability and satisfaction, which stood out when reviewing step flow discovery center – The step-based browsing is clear, and product discovery feels simple and well organized.
During a routine browsing session, I came upon view this shop and realized that the site flows well, with items arranged in a way that improves navigation.
As I reviewed various online marketplaces, I came upon this tidy shopping site and found the user experience enjoyable due to its clear organization and smooth navigation between different product categories.
Individuals passionate about modern fashion often explore websites that present curated clothing lines featuring aesthetic inspired designs and stylish wearable pieces Aesthetic Wardrobe Boutique – showcasing a fashion platform that highlights modern clothing collections focused on elegance simplicity and trend aligned designs created for individuals who value expressive yet minimal personal style choices
Online shoppers who enjoy creative digital marketplaces often appreciate platforms designed with a trading post theme where product browsing feels refreshing and unique due to its structured yet exploratory layout that encourages discovery across different categories in an engaging way Quartz Orchard Trading Post Hub – offering a trading post inspired shopping experience where unique product presentation creates a refreshing browsing journey with organized sections designed to make exploration easy enjoyable and visually engaging for everyday users
During my search for online deals, I explored visit this trend listing and saw trendy products that make the site feel like a fun place to shop for stylish items.
Many shoppers today look for reliable online stores and while browsing different options you may notice useful deals where budget friendly shop portal appears within trusted recommendations offering reasonably priced everyday essentials and quick shipping options for customers seeking convenience and value across various categories online.
Users interested in exploring online retail options often look for websites that combine variety with simple navigation and clear product arrangement retail deck explorer – The platform presents items in an easy to follow format, making it convenient for shoppers to compare products and move through categories without feeling overwhelmed during browsing sessions
People who shop online regularly often choose websites that offer minimal design and easy navigation flow where easy deals shopping center is featured in content and it highlights a system designed to improve browsing flow while ensuring users can quickly access products and complete purchases smoothly across all available categories without confusion.
While analyzing ecommerce platforms designed for digital and tech-savvy audiences, I observed that modern shopping points enhance usability through organized presentation of gadgets and tools, which became evident when testing digital product discovery hub – The site feels sleek and modern, showcasing tech-focused items that appear appealing and relevant to today’s users.
During my review of outdoor travel sites, I found this camping activity page and thought it was a fun and useful resource to browse, with helpful ideas that make outdoor planning feel easier and more enjoyable.
While exploring various gaming hubs, I came across this gaming content platform page and gaming content seems fun, modern, and fairly engaging to explore, with a structure that feels dynamic and well designed.
Modern consumers often expect digital shopping platforms to deliver both speed and clarity, ensuring that each interaction feels immediate and the overall process remains easy to follow from start to finish efficient shopping portal – the experience is commonly described as well-optimized, supporting quick navigation and reducing delays during checkout steps.
While analyzing ecommerce systems focused on cart efficiency and speed, I encountered a section called fast purchase cart index – The platform delivers a smooth browsing experience where cart actions respond instantly, helping users complete shopping steps quickly and efficiently without unnecessary waiting or delays.
На платформе https://android-carplayers.ru/ очень прозрачная система выплат, деньги приходят на карту довольно быстро.
In the process of evaluating digital fashion stores focused on style and affordability, I found that structured deal hubs enhance user experience and engagement, which became evident when reviewing fashion style discount hub – The fashion section feels modern, and the deals look trendy, attractive, and well organized for browsing.
продвижение сайтов в москве яндекс гугл [url=https://stars-top.ru/effektivnoe-prodvizhenie-sajtov-v-moskve-strategii-dlya-liderstva/]продвижение сайтов в москве яндекс гугл[/url]
seo специалист услуги [url=https://newsomsk.ru/news/152386-seo_moskva_realny_keys_rosta_lokalnogo_brenda_s_ud/]seo специалист услуги[/url]
сравнение сайта в москве [url=https://dettka.com/kakimi-metrikami-izmerit-effektivnost-digital%e2%80%91strategii-dlya-stolichnogo-biznesa/]сравнение сайта в москве[/url]
раскрутка сайта москва поисковое продвижение [url=https://lezgigazet.ru/archives/406934]раскрутка сайта москва поисковое продвижение[/url]
During my exploration of e-commerce sites, I noticed this everyday purchase page and it appears to offer a clean and simple structure, helping users find common products with minimal effort.
разработка продвижение и сопровождение сайтов [url=https://interviewrussia.ru/interesno-obo-vsyom/audit-sajta-czena-ot-chego-zavisit-stoimost-tehnicheskogo-analiza-i-pochemu-ne-stoit-ekonomit/]разработка продвижение и сопровождение сайтов[/url]
While browsing casually through different platforms, I came across browse trusted auto hub and it felt reliable, since I experienced no issues while navigating through the pages.
Fashion oriented users often seek brands that highlight stylish and minimalist clothing collections designed with modern aesthetic influences for everyday outfits Stylish Aesthetic Apparel Hub – offering a curated fashion platform featuring elegant clothing collections and contemporary designs that focus on comfort style and versatility for individuals who appreciate modern wardrobe aesthetics
Shoppers searching for reliable e-commerce platforms usually prefer websites that emphasize both value and simplicity in browsing where affordable cart options center is included in guides and it highlights a marketplace designed to offer competitive prices while maintaining a smooth and user friendly shopping experience for everyday buyers.
In the middle of checking different eCommerce sites, I paused at take a look way store and found the shopping flow very simple, with a user friendly layout that makes browsing smooth and effortless.
In the course of evaluating online retail platforms focused on user journey optimization, I found that flow-based navigation enhances usability by creating logical browsing paths, which was evident when analyzing guided shopping flow hub – The platform provides smooth transitions across sections, making the overall experience feel intuitive and well connected.
Many online visitors exploring structured ecommerce platforms often highlight the importance of guided navigation, especially when using navigation deals center – Users can easily follow categorized paths that make browsing deals more straightforward and reduce time spent searching for relevant items.
Individuals who enjoy calm digital environments often prefer platforms that use lounge themes to create visually soothing browsing experiences with well structured product layouts Velvet Valley Serene Lounge Hub – featuring a clean online marketplace where lounge inspired aesthetics enhance product presentation and create a smooth browsing experience designed for comfort and visual appeal
Shoppers searching for reliable online marketplaces usually prefer websites that combine affordability with daily updated deals where affordable daily shopping portal is featured in guides and it highlights a system designed to make online shopping easier while ensuring users can efficiently browse products and enjoy a seamless checkout experience overall.
While conducting comparative UX analysis of modern shopping systems, I came across wave commerce listing center – The platform features a clean wave themed layout that enhances structure, improves navigation clarity, and supports efficient browsing across multiple product categories in a seamless way.
As I continued checking healthcare-related websites, I ran into this beauty clinic site and found the layout very user-friendly, with smooth navigation that made the browsing experience easy, clean, and comfortable overall.
In the process of browsing news update sources, I came across this Fast With Gaza updates hub and content appears focused on updates, presented in simple readable format, with a layout that emphasizes clarity and simplicity.
While analyzing ecommerce usability patterns across different digital storefronts, I found that cart efficiency and system stability significantly impact user confidence, especially when browsing large product catalogs, which became apparent when reviewing stable checkout cart – The shopping experience feels consistent and reliable, with a cart system designed to keep interactions simple, responsive, and easy to manage throughout the entire purchase journey.
In the process of analyzing digital commerce platforms focused on cart usability and switching ease, I found that flexible cart structures enhance shopping experience and efficiency, which became evident when exploring simple cart flow hub – The system allows users to switch products easily, keeping the shopping process smooth and clear.
During my search for online bargain resources, I encountered this savings corner link and it appears to offer useful deals that are worth revisiting periodically to catch fresh discounts and special offers.
While scanning through online stores for something new, I stopped at tap to open and found the layout clean with enough variety to consider coming back again later.
Many shoppers looking for convenient digital marketplaces often choose platforms that simplify browsing and reduce unnecessary steps where quick access shopping hub appears in content describing efficiency – it highlights a system designed to improve speed and help users complete purchases smoothly without confusion or interruptions during the process.
Individuals exploring new fashion ideas often prefer online platforms that showcase modern clothing with aesthetic inspired designs suitable for everyday styling Modern Elegance Fashion Hub – presenting a curated clothing platform that emphasizes stylish apparel aesthetic design principles and wearable fashion collections tailored for individuals seeking balanced and contemporary wardrobe inspiration
many online shoppers exploring general ecommerce platforms often appreciate websites that combine variety with simple navigation making it easier to browse multiple product categories without confusion or delays during everyday shopping sessions total shop hub explorer often described as a convenient marketplace – the platform provides a wide selection of products while maintaining a clean and structured browsing experience that helps users find items quickly and comfortably across all categories available
People who enjoy organized digital stores often prefer platforms that use merchant lane systems to present products in clear structured pathways for easier browsing and discovery Harbor Jasper Navigation Goods Hub – featuring a streamlined shopping experience where products are organized through a merchant lane layout designed to improve clarity and enhance user experience across all product categories
goodsmixstore.shop – Mixed goods selection is useful, plenty of options in one store
Many consumers browsing online stores often appreciate platforms that highlight secure payments and trusted shopping carts where easy cart trust portal is included in guides and it highlights a system designed to improve usability while ensuring users can quickly find products and complete transactions without unnecessary effort or confusion.
During a comparative study of online marketplaces focused on structured navigation and product accessibility, I discovered that category flexibility enhances usability significantly, especially in large scale catalogs, which became evident when reviewing smart switch category portal – The platform offers variety and allows users to move easily between product categories, ensuring a smooth and efficient browsing journey overall.
While exploring illustrated game and puzzle collections, I noticed visual puzzle art collection and puzzles look creative, colorful, and quite enjoyable for visitors overall, presenting designs that feel imaginative, high quality, and engaging for anyone interested in artistic leisure activities. – The overall feel is light and enjoyable.
While exploring online mom communities, I came across this Chicago mom lifestyle site and found the topics quite relatable today, offering a realistic look into everyday parenting experiences.
While reviewing ecommerce platforms designed for smooth and fast browsing, I noticed that shopping zones improve user experience and reduce delays, which stood out when exploring speedy shopping experience center – The shopping zone is fast, and all pages load smoothly with excellent responsiveness.
While reviewing different shopping websites, I found this intuitive shopping link and liked how everything is arranged logically, making product discovery quick and effortless without having to dig through complicated menus.
Online buyers who prioritize security tend to look for platforms that offer trusted seller connections and reliable order handling where safe trade cart hub is featured in guides – it reflects a marketplace built to support secure transactions while ensuring users can shop easily and confidently without worrying about product authenticity or payment safety.
People interested in curated fashion often explore online stores that offer aesthetic driven clothing collections combining simplicity with modern design trends Modern Outfit Inspiration Hub – providing a stylish fashion destination that showcases clothing collections focused on aesthetic appeal contemporary design and wearable comfort for individuals seeking fresh and expressive wardrobe ideas
While exploring online marketplaces, I stopped at explore value cart site and found many budget friendly choices, so I would likely check it again in the future.
seo продвижение в москве в топ [url=https://golubevod.net/seo-agentstvo-moskva.html]seo продвижение в москве в топ[/url]
частный seo специалист [url=https://astera.ru/articles/seo-prodvizhenie-agentstvo/]частный seo специалист[/url]
While exploring multiple online marketplaces focused on pricing and product variety, I came across a section labeled tech deals browsing hub – The platform presents digital and tech products at appealing prices, making it feel like a convenient place for users looking for budget friendly online shopping options overall.
People who value easy online shopping often look for outlet stores that provide organized categories and a decent range of practical products in a simple layout Cloud Cove Outlet Essentials Store – offering a streamlined shopping experience focused on simplicity and convenience where users can browse items easily and find useful products without unnecessary complexity in the browsing process
Many consumers exploring online shopping options frequently choose platforms that offer streamlined browsing and checkout processes where quick shopping access hub is included in descriptions and it highlights a system designed to improve efficiency while ensuring users can easily find products and complete transactions with minimal effort and maximum convenience.
While checking community gathering information platforms, I encountered iftar outreach community site and information here seems community focused, helpful, and well structured content, with a presentation that feels welcoming and informative for general visitors. – The layout feels approachable and clear.
As I continued browsing seasonal event platforms, I came across this festive celebration site and it feels like a nicely put together site with good information, making the browsing experience smooth and informative from start to finish.
During analysis of ecommerce systems emphasizing cart zones and checkout simplicity, I discovered that structured design improves usability and trust, which became clear when exploring efficient checkout cart zone portal – The cart zone is clean, and checkout is simple, quick, and easy to complete.
During my review of various support-focused platforms, I encountered a comforting resource and found the writing style to be very considerate, offering guidance in a way that feels balanced, empathetic, and easy for readers to follow without confusion.
Online consumers who value structured shopping environments frequently engage with systems that improve clarity when they use unified cart marketplace while exploring deals – It focuses on bringing multiple product types into one accessible interface to enhance usability and simplify decision making.
People interested in curated fashion often explore online stores that offer aesthetic driven clothing collections combining simplicity with modern design trends Modern Outfit Inspiration Hub – providing a stylish fashion destination that showcases clothing collections focused on aesthetic appeal contemporary design and wearable comfort for individuals seeking fresh and expressive wardrobe ideas
While exploring football event tracking websites, I came across global tournament scoreboard page and sports related updates feel exciting, timely, and easy to follow, presenting match data in a way that feels simple, direct, and easy to understand. – It feels practical and fan-friendly.
Shoppers exploring e-commerce platforms frequently prefer websites that focus on secure checkout and clean cart design where smart safe cart portal appears in descriptions and it reflects a system designed to reduce complexity while helping users easily browse products and enjoy a smooth and efficient online shopping experience overall.
As I continued browsing online marketplaces, I discovered this smart product page and liked how the structure makes finding items effortless, with a clean and logical design that improves the overall shopping experience.
Shoppers who appreciate stylish online platforms often prefer websites that use market studio concepts to make browsing more visually engaging while keeping product organization simple and clear Kettle Willow Market Studio Design Hub – offering a refined e commerce experience where creative studio inspired layouts enhance product presentation making online shopping more enjoyable and visually appealing for everyday users
While casually exploring various platforms, I found explore this site – The initial feel is quite positive, as the design seems smooth, balanced, and comfortable enough to encourage further browsing without hesitation.
продвижение раскрутка сайта москва александр [url=https://in.gallerix.ru/journal/internet/201811/zakazat-raskrutku-sayta-v-moskve-kak-vybrat-nadezhnogo-podryadchika-bez-finansovyx-riskov/]продвижение раскрутка сайта москва александр[/url]
People looking for convenient shopping options often prefer platforms that centralize essential products effectively where simple essentials network is included in descriptions highlighting usability – it supports quick browsing and efficient ordering for everyday items while maintaining a clean and user friendly digital shopping environment.
As I browsed through several informative platforms, I noticed this practical resource and found that it delivers its niche content in a way that feels both educational and enjoyable to read.
Fashion lovers often explore online brands that present modern clothing collections inspired by aesthetic design and contemporary fashion trends for versatile styling Elegant Apparel Fashion Studio – showcasing a curated platform of stylish clothing and aesthetic focused collections designed to support modern wardrobe needs while emphasizing creativity comfort and refined personal expression
While reviewing multiple online resources, I stumbled upon explore here – The design looks clean and modern, and everything feels smooth and well organized while browsing through pages and sections.
While going through abstract conceptual frameworks online, I came across chaos systems reflection site and unique concept presented here, thought provoking and quite intriguing overall, presenting ideas that feel layered, philosophical, and open to interpretation. – The experience feels deep and intellectually engaging.
Shoppers who value speed and simplicity in online purchasing often explore platforms designed to centralize essential goods and everyday items daily essentials cart hub providing a straightforward way to access useful household products while saving time during routine buying activities.
In the process of checking clothing websites, I encountered this diverse wardrobe hub and it looks like a solid place to check out different styles, offering fashion choices that appear suitable for different tastes and preferences.
Shoppers searching for reliable e-commerce platforms often value websites that provide secure cart solutions and simple navigation where smart secure cart center appears in content and it reflects a system designed to enhance usability while helping users quickly browse products and complete purchases without unnecessary complications or confusion in the process.
Individuals who appreciate thoughtfully organized online stores often look for platforms that use boutique hub designs to group products according to different customer needs and preferences Grove Harbor Everyday Boutique Hub – featuring a structured e commerce platform where curated items are displayed in a clean layout designed to improve browsing clarity and help users find products quickly and efficiently
In the process of analyzing digital commerce platforms focused on minimalism and usability, I found that basic layouts enhance browsing satisfaction and clarity, which became evident when exploring simple store browsing hub – The design looks clean and organized, making it easy to explore different products in a comfortable way.
As I explored different websites focused on creative services, I ran into this unique resource and it impressed me with how it blends originality with functionality, making it feel like a useful destination for those seeking fresh ideas.
While navigating through various online resources, I encountered see more here – I took a quick look and the layout appears clean and organized, offering a smooth browsing experience overall.
People who enjoy aesthetic fashion often look for curated clothing brands that combine modern design elements with stylish and versatile apparel collections Modern Style Apparel Hub – offering a fashion platform that features elegant clothing collections inspired by contemporary aesthetics and designed for individuals seeking comfortable stylish and expressive wardrobe options for everyday use
As I continued reviewing online research materials, I noticed clinical information page and the layout makes everything feel very accessible, allowing readers to follow the details easily without struggling with complicated structure or unclear explanations.
Online buyers who value convenience often look for platforms that offer organized listings and easy checkout flow where smart deal nest center is included in descriptions and it highlights a system designed to enhance usability while ensuring users can quickly browse products and complete purchases with minimal effort and maximum efficiency.
besttrendstation.shop – Easy to navigate site, everything feels structured and user friendly.
Wow! After all I got a weblog from where I be able to really take useful information concerning my study and knowledge.|
While analyzing various ecommerce platforms focused on structured visual design and usability, I noticed that grid-based layouts often improve product discovery and make browsing more efficient for users, especially in large catalogs, which became clear when reviewing smart grid shopping hub – The grid layout feels clean and organized, with products arranged in a very easy to understand structure that helps users browse smoothly without confusion or unnecessary visual clutter.
While reviewing ecommerce platforms focused on streamlined navigation and product discovery, I observed that smooth interfaces improve usability, which was clear when testing modern shopping network portal – The marketplace feels smooth, and browsing products is quick, simple, and intuitive.
During some light online research, I ran into view this link – The site is simple in design, everything loads quickly, and it feels easy to navigate without unnecessary complexity.
Online consumers often prefer shopping environments that combine speed, clarity, and accessibility in one place where fast checkout cart point appears in guides – it represents a streamlined system that allows users to complete purchases quickly while maintaining a simple and stress free browsing experience throughout their shopping journey.
In the process of browsing relaxing websites, I came across this Elphin Bothy overview page and the site feels calm, informative, and very pleasant to browse through, with a layout that feels soft, simple, and easy to follow.
In the process of evaluating digital commerce platforms focused on trust and usability, I found that trusted buying hubs enhance browsing flow and satisfaction, which became evident when reviewing secure shopping experience portal – The site feels reliable, and navigation is clean, smooth, and easy to understand.
https://t.me/Asiapsi
As I browsed through various music and entertainment platforms, I noticed this artist showcase site and found the content pretty engaging, which kept me browsing for a while as I explored the different sections and offerings presented.
People who appreciate aesthetic fashion often search for brands that offer curated clothing collections focused on modern simplicity and expressive personal style Modern Apparel Collection Hub – featuring a fashion platform dedicated to stylish clothing and aesthetic inspired designs that blend elegance and comfort while offering versatile wardrobe pieces for individuals with a taste for contemporary trends
People who enjoy structured but energetic e commerce platforms often look for websites that use trading post themes to create dynamic browsing flows with varied product selections that improve usability and enjoyment Wave Harbor Exchange Trading Hub – providing a clean marketplace where trading post styling enhances product display and ensures a dynamic browsing experience that feels smooth and engaging across all shopping sections
While checking out different online resources, I stumbled upon click here now – It came across as an interesting site, and navigating through its pages felt smooth, simple, and fairly effortless overall.
Many shoppers comparing digital marketplaces often pay attention to how quickly pages load and how intuitive navigation feels on CleverCart Arena Digital Bazaar – Consistent styling and clear section hierarchy make the browsing process smoother and more enjoyable for returning visitors overall.
While browsing regional information platforms, I noticed this warm community page and liked how it presents content in a friendly and inviting way that makes visitors feel immediately at ease.
In the process of evaluating digital retail environments focused on smart purchasing systems, I found that structured buying concepts improve user satisfaction by making navigation more predictable and easy to follow, which was clear when reviewing smart shopping experience index – The platform provides smooth and simple navigation that helps users browse products easily without unnecessary steps or confusion.
In analyzing multiple online product catalog systems, I observed a design centered around WinterPeak Vendor Hub which ensures content is grouped logically and displayed with consistent styling for better usability – the browsing experience feels fluid and users can move between sections without confusion.
In the course of evaluating online shopping platforms centered around deals and discounts, I found that clarity in pricing improves user confidence and engagement, which was evident when exploring daily deals showcase portal – The deals section looks attractive, and prices seem reasonable, structured, and easy to compare.
People looking for efficient online shopping experiences frequently prefer platforms that highlight stylish design and smooth navigation where ultimate urban shopping hub is featured in content and it highlights a system designed to improve usability while ensuring users can quickly discover products and enjoy a seamless and satisfying shopping experience overall.
Many digital shoppers prefer websites that offer warm visual design and intuitive navigation especially when accessing Meadow Cotton Goods Store – The site feels cozy and welcoming making it very easy to browse through different sections without confusion or unnecessary effort during shopping.
During a casual browsing session across various websites, I noticed visit this link – It has a nice overall structure, and the calm tidy browsing experience makes navigation feel smooth and easy to follow.
While analyzing ecommerce UX designs centered on modern cart systems and clean presentation, I observed that polished layouts improve browsing comfort and satisfaction, which was evident when reviewing modern cart interface hub – The design feels refined and minimal, offering a seamless shopping experience that feels effortless.
Online buyers often look for platforms that ensure consistent updates in pricing and product availability across categories where loop based savings store is mentioned in content describing flexible shopping systems – it highlights a model built to deliver ongoing value through changing deals and refreshed product listings.
In the middle of browsing informational websites, I encountered this Elkhart initiative project page and project information seems structured, useful, and clearly presented overall today, offering a layout that feels intuitive and easy to navigate.
Individuals passionate about modern fashion often explore websites that present curated clothing lines featuring aesthetic inspired designs and stylish wearable pieces Aesthetic Wardrobe Boutique – showcasing a fashion platform that highlights modern clothing collections focused on elegance simplicity and trend aligned designs created for individuals who value expressive yet minimal personal style choices
As I explored different online group platforms, I noticed this community hub link and appreciated that it is still active and consistently updated, which makes it feel relevant and engaging for returning visitors.
People who enjoy structured online retail experiences often look for platforms that present products in merchant mart layouts with well defined categories for improved usability and browsing speed Canyon Goods Icicle Mart Space – providing a clean and organized shopping platform where products are grouped into categories using a merchant mart concept designed to enhance navigation and make online shopping more efficient and user friendly
Digital marketplaces are shifting toward more user-friendly systems that prioritize speed, accessibility, and real-time updates for all promotional listings Quick Price Drop Center – The platform ensures users are informed about the latest reductions, helping them take advantage of savings before offers expire or change
As I explored different awareness-driven websites, I came upon this engaging example and found that the content is both meaningful and well delivered, making it easy to follow and understand.
During a casual search for interesting resources, I ran into visit this link – The organization feels very user-friendly, allowing visitors to quickly understand where things are and explore content smoothly without dealing with cluttered or confusing layouts.
In today’s competitive online retail landscape, smooth user experience and structured design are crucial when visiting sites such as CartArena Explorer CleverCart Space – A well organized layout helps reduce cognitive load and allows users to focus more easily on browsing products and content.
During analysis of online retail systems focused on competitive pricing and value perception, I discovered that affordable platforms enhance user trust by offering consistent deals, which became clear when testing budget product discovery portal – The pricing appears fair and attractive, making it feel like a reliable place for budget friendly shopping.
While evaluating modern shopping platforms with simplified structures, I noticed improved usability when interacting with systems like easy order cart – The system is designed so users can manage items effortlessly, making the entire shopping process smooth and beginner friendly.
While analyzing ecommerce websites focused on electronics and gadget offers, I encountered a section called digital tech deals index – The layout presents tech products in a clean and organized way, making it easy for users to browse gadgets that appear useful, modern, and reasonably priced for general consumers.
While analyzing different e-commerce style platforms, reviewers often emphasize the role of well-organized collections, and within that context the embedded link Market Guild Explorer acts as a helpful gateway for users seeking structured browsing experiences and better insight into available marketplace categories.
Many online shoppers look for convenient platforms that combine variety and affordability when browsing everyday essentials so they often land on pages that include daily essentials hub while exploring product options and then continue reading reviews – the store presents a broad mix of household items and useful goods designed for routine purchasing needs across different categories and user preferences.
Many shoppers prefer platforms that offer stress free browsing and clean layouts especially when they visit Meadow Cotton Shop Outlet – The site feels welcoming and well structured making browsing easy comfortable and enjoyable for users exploring different products.
During a quick search session, I noticed this useful link – It has an interesting design approach, and the pages are clear and not overwhelming at all, which helps create a smooth experience.
Shoppers searching for reliable e-commerce platforms often value websites that provide speed and simplicity in browsing where quick fast shopping hub appears in content and it reflects a system designed to enhance usability while helping users quickly browse products and complete purchases without unnecessary complications or confusion in the process.
While checking analytical writing platforms, I noticed deep insight reflection site and content feels reflective, detailed, and somewhat thought provoking overall tone, presenting ideas in a way that feels methodical, structured, and intellectually engaging. – It has a quiet sense of depth and clarity.
Fashion enthusiasts looking for stylish clothing often prefer brands that offer curated aesthetic collections reflecting modern trends and versatile design approaches Contemporary Wardrobe Fashion Hub – offering a curated fashion platform featuring modern clothing collections and aesthetic inspired designs that emphasize elegance versatility and comfort for individuals seeking refined and expressive personal style choices
Online users often appreciate e-commerce platforms that provide clarity and trend focus especially when accessing Digital Hub Station Trends It is a nice platform for trends where products feel updated and quite useful making browsing simple and effective across all sections.
As I continued checking game-related websites, I discovered this singles interactive game page and it seems fun and unique compared to typical websites online, with a concept that feels different and more experimental than usual.
Let’s face it: PFL Sioux Falls is essentially a homecoming event for Logan Storley packaged as a legitimate MMA event.
Individuals who enjoy boutique style shopping online often seek platforms that combine creativity with functionality offering product listings that feel more like curated exhibitions than standard catalogs Orchard Atelier Design Hub – providing a refined online store experience that emphasizes artistic product presentation and atelier inspired styling designed to make browsing more visually appealing and enjoyable for users who appreciate creative digital spaces
As I continued browsing luxury travel accommodations, I ran into this refined lodge showcase and found the design very appealing, with a calming visual style that strongly reflects a premium and peaceful retreat experience.
While reviewing ecommerce platforms designed for open structure and category navigation, I noticed that spacious layouts improve user comfort and discovery, which stood out when exploring open browse category center – The interface appears airy and uncluttered, making it easy to explore different sections.
The Sanford Pentagon in South Dakota somehow manages to turning intense combat come across as strangely close-up.
While casually exploring online platforms, I stumbled upon view this link – At first glance, it looks well put together, offering a streamlined interface that feels easy to understand and pleasant to interact with.
While analyzing ecommerce UX designs centered on cart functionality and efficiency, I observed that direct cart hubs improve user satisfaction and reduce complexity, which was evident when reviewing smart purchase cart center – The platform delivers a fast and clean checkout flow that feels intuitive and smooth.
Many digital storefronts focus on reducing friction and improving user experience through better design CleverCart Flow Market – Browsing feels natural and well structured across the site Clear categorization ensures users can quickly locate items without unnecessary effort or delay in process flow maintained.
Many users exploring e-commerce systems appreciate platforms that focus on connecting them with verified sellers in a structured environment where easy access link marketplace appears in content highlighting convenience – it emphasizes a smooth shopping experience designed to make browsing faster while ensuring reliable access to various product categories and deals.
Online shoppers often value platforms that deliver structured design and fast performance especially when accessing Goods Berry Cove Online Hub – I enjoyed browsing because everything feels nicely arranged and the interface makes shopping simple and easy to follow.
While reviewing online retail platforms for design simplicity, I discovered a listing titled easy access shop gate – The structure is minimal and user friendly, ensuring quick product discovery and smooth navigation through categories without unnecessary steps or visual clutter affecting usability at any stage.
When evaluating digital commerce platforms, usability is a common focus, and Brook Vendor Showcase – The system performs reliably with quick response times and well-organized content blocks that help users navigate through listings efficiently while maintaining a clean and accessible interface.
https://telegra.ph/ACA-203-Betting-Preview-Gaforov-vs-Akopyan-04-29
Absolute Championship Akhmat 203 comes to Tashkent with a high-stakes flyweight headliner and a fight lineup that draws strongly from the promotion’s strong presence across Uzbekistan.
rent a car Tivat fleet car hire Tivat
As I was checking different platforms online, I discovered open this page – Everything is arranged in a clean way, and it makes finding items effortless while keeping browsing simple and comfortable.
People who enjoy online shopping often choose platforms that combine simple navigation with quick buying options where quick shopping zone hub appears in content and it reflects a system built to enhance browsing efficiency while helping users easily discover products and complete transactions smoothly across various everyday shopping categories.
Individuals passionate about modern fashion often explore websites that present curated clothing lines featuring aesthetic inspired designs and stylish wearable pieces Aesthetic Wardrobe Boutique – showcasing a fashion platform that highlights modern clothing collections focused on elegance simplicity and trend aligned designs created for individuals who value expressive yet minimal personal style choices
UFC statistics
Headlining the UFC PFL Sioux Falls card Logan Storley faces the reigning 2024 PFL Europe champion Florim Zendeli.
As I continued exploring event guides online, I noticed this Edinburgh Big Day Out listing site and event information looks lively, well organized, and easy to understand, offering a smooth browsing experience with clear and structured information.
Online users often prefer e-commerce websites that focus on speed and efficiency especially when they access Zone Dynamic Cart Market Shopping experience feels smooth and the cart system works really fast today allowing users to enjoy quick actions without unnecessary loading delays.
As I continued exploring online travel resources, I came across this local Ireland info page and found it helpful, especially for someone new to this topic, offering straightforward and beginner-friendly content throughout.
E-commerce sites that prioritize streamlined navigation and logical category grouping tend to offer better user experiences and higher satisfaction among returning customers, and one example is ShopStream Online – The platform focuses on delivering a continuous flow of browsing that supports fast decision-making and efficient product discovery for all types of users.
While reviewing ecommerce systems designed for clarity and minimalism, I observed that fresh hub layouts improve product visibility and navigation flow, which was evident when testing fresh browsing experience portal – The interface feels simple and organized, allowing browsing to remain easy and comfortable.
While browsing through curated design collections, I found an appealing resource that showcases a refined approach, with clean lines and thoughtful spacing that contribute to a professional and attractive overall look.
People who enjoy practical online stores often prefer websites that organize products in a district format to simplify browsing and improve shopping flow Golden Cove Easy District Hub – providing a structured e commerce platform focused on goods district organization that helps users navigate products quickly and enjoy a smooth and efficient online shopping experience
People browsing e-commerce platforms often prefer systems that improve efficiency by organizing seller connections clearly where safe purchase link center is included in descriptions – it highlights a structured marketplace designed to support smooth transactions while helping users find reliable products and complete orders without unnecessary delays.
In the process of evaluating digital commerce platforms focused on structured product organization, I found that organized marketplaces enhance usability and browsing efficiency, which became evident when reviewing efficient buying hub center – The platform is cleanly structured, making it easy to find products in a short amount of time.
As I explored different websites, I came across learn more here – It appears useful overall, with a clear presentation style that allows users to browse and understand content without confusion or difficulty.
During a comparative study of online retail systems emphasizing category consolidation and usability, I discovered that ultra hub platforms help users access diverse products without switching between multiple sites, which stood out when exploring central ultra marketplace portal – The platform feels modern and flexible, with lots of categories available in one place, making browsing smooth, structured, and user friendly overall.
Many users browsing online stores prefer simple layouts and efficient navigation especially when accessing Silk Goods Atelier Ridge The interface feels clean and smooth navigation made this a pleasant visit ensuring a comfortable and easy browsing experience across all sections of the site.
E-commerce users frequently prefer sites that balance design and functionality, especially when interacting with Clever Shopping Zone – The interface provides a calm browsing flow, fast loading elements, and clear organization that enhances overall usability and user satisfaction across sessions.
When evaluating modern e-commerce platforms and digital vendor ecosystems, many users emphasize structural clarity, and Cove Shopping Atelier Network is often cited in discussions about well-organized marketplaces – the browsing experience feels consistent and intuitive, allowing users to move between sections without confusion or unnecessary interruption during navigation.
People interested in refined fashion often browse websites that feature aesthetic clothing collections designed to match modern style trends and personal expression Chic Fashion Collection Space – offering a curated selection of stylish apparel focused on modern aesthetics and versatile wardrobe pieces that combine elegance comfort and trend driven design for contemporary fashion lovers
During some light online research, I ran into view this link – The interface feels smooth and well built, and it is easy to explore content while maintaining a clear and simple layout.
In the middle of browsing advocacy websites, I encountered this Florida civic improvement site and the website presents optimistic messaging, clear goals, and simple layout design, offering structured information that feels simple and readable.
Online shoppers often look for platforms that highlight bulk deals and affordable product selections where budget buy deals hub appears in listings and it reflects a system designed to help users discover more savings while browsing a wide range of everyday products with smooth navigation and easy checkout experiences across all categories.
eva casino
In online shopping environments users frequently prioritize good pricing and reliable deals especially when visiting Dynamic Center Deal Zone Deals here are attractive and prices seem fair and competitive online allowing users to browse confidently and compare offers without confusion or unnecessary complexity.
During my browsing of modern web examples, I discovered this minimalist showcase page and I like the overall presentation, feels modern and straightforward, offering a smooth and visually calm experience overall.
While analyzing ecommerce platforms designed for clarity and efficient browsing, I observed that core layouts improve navigation by presenting products in a readable format, which became evident when testing easy read product center – The layout feels simple and organized, allowing users to browse listings that are clear and easy to follow.
In the middle of reviewing various public-facing platforms, I came upon a straightforward website that clearly prioritizes readability, making the overall message easy to follow from start to finish without unnecessary complications.
Many consumers exploring online stores appreciate marketplaces that support fast decision making and seamless checkout processes where instant shopping cart hub appears in informational content – it represents a streamlined shopping experience that allows users to quickly browse items and complete purchases efficiently with minimal effort required.
Online users who prefer organized digital marketplaces often appreciate platforms designed as commerce hubs where a wide selection of products is available with smooth navigation making browsing efficient and enjoyable across multiple categories in a structured online environment Velvet Ridge Commerce Central Hub – offering a well structured commerce hub experience where wide product selection is paired with smooth navigation helping users browse categories easily and efficiently in a clean and intuitive online shopping environment
While conducting usability research on ecommerce systems focused on essential goods, I noticed that daily needs stores improve accessibility and shopping comfort, which became evident when analyzing organized essentials shopping hub – The site is practical and well structured, making everyday items easy to find and purchase.
During usability testing of ecommerce platforms focused on decision freedom, I came across a section titled product flexibility shopping hub – The interface is structured to encourage easy selection, making browsing simple and allowing users to choose products freely without confusion or unnecessary interface complexity.
Shoppers frequently appreciate platforms that offer clean layouts and intuitive navigation especially when they visit Grove Guild Retail Raven Hub The structure is interesting and user friendly making browsing comfortable and helping users quickly understand where everything is located.
Fashion enthusiasts exploring modern style often look for online brands that combine contemporary aesthetics with wearable clothing collections designed for everyday elegance and confidence in personal expression Modern Style Fashion Hub – offering a curated fashion platform featuring stylish apparel and aesthetic driven collections that reflect current trends while emphasizing individuality comfort and refined design for a modern lifestyle audience
In the middle of checking various resources, I discovered check out this page – After trying the site, I found navigation to be smooth and reliable, with no issues while browsing through its content.
When analyzing online shopping behavior trends, researchers often note that users prefer platforms that reduce cognitive load through clear design and predictable navigation structures across all pages Harbor Stone Vendor Flow – the browsing experience is described as smooth and consistent, with minimal effort required to move between categories and evaluate product differences.
While browsing through several online options, I came across take a look here – The pages load fast, and the experience feels responsive and user friendly, making everything quick and easy to explore.
Online browsing becomes more enjoyable when websites offer clean layouts and fast interaction, especially when users access Clever Deals Navigator – The interface helps guide users naturally through different categories, making it easier to find relevant deals without feeling overwhelmed or distracted.
продвижение молодого сайта в москва [url=https://prostudio.ru/journal/pretty/kak-vyvesti-sajt-v-top-po-moskve-prakticheskaya-seo-strategiya/]продвижение молодого сайта в москва[/url]
While surfing through unconventional content platforms and amusing web collections, I discovered lighthearted Fred Thompson corner and the name alone sets a comedic tone, while the content itself feels relaxed, mildly absurd, and designed to keep visitors casually entertained without any pressure or seriousness. – Definitely has a quirky charm that stands out quickly.
Shoppers searching for reliable e-commerce platforms often value websites that provide wide selections and structured navigation where smart world shopping portal appears in content and it reflects a system designed to enhance usability while helping users quickly browse products and complete purchases without unnecessary complications or confusion in the process.
Online visitors exploring a wide range of consumer goods often come across listings that include shopping browse section within their search results helping them refine choices and evaluate different product types more comfortably especially during extended browsing sessions online experiences – Product organization supports smooth exploration without overwhelming the user.
While reviewing ecommerce systems designed for organized browsing and structured layouts, I observed that line-based shop systems improve navigation and clarity, which was evident when testing linear shopping experience hub – The interface feels neat and easy to follow, helping users locate products quickly.
As I continued browsing regional content hubs, I noticed this southwest coast exploration page and it seems like a niche site but still quite informative overall, providing focused information in a clean and understandable layout.
People shopping online often value platforms that combine wide product variety with easy usability, and this is demonstrated in Global Dock Shop – The service offers a user centered experience that simplifies browsing and helps customers find suitable items across multiple categories with minimal effort and maximum convenience.
In the process of exploring luxury product websites, I came across this stylish jewelry hub and it impressed me with its clean organization and sophisticated design that enhances the overall perception of the collection.
People looking for affordable electronics accessories usually prefer platforms that provide variety and clear navigation where t modern tech savings hub helps highlight budget friendly deals and ensures users can find useful gadgets quickly while enjoying a practical and efficient online shopping environment for daily device upgrades.
While analyzing ecommerce platforms optimized for fast and direct shopping experiences, I noticed that simplified flows improve user satisfaction and transaction speed, which stood out when reviewing quick direct buy hub – The navigation is straightforward and the entire purchasing process feels smooth and efficient.
In the course of reviewing online retail systems focused on speed optimization, I observed a section labeled quick response shopping center – The platform delivers a seamless experience where pages load instantly and navigation feels fluid, helping users browse products efficiently and without frustration.
Как технические ошибки на сайте мешают [url=https://moypodolsk.crforum.ru/viewtopic.php?t=2209]Поисковое продвижение сайта[/url]?
Individuals exploring new fashion ideas often prefer online platforms that showcase modern clothing with aesthetic inspired designs suitable for everyday styling Modern Elegance Fashion Hub – presenting a curated clothing platform that emphasizes stylish apparel aesthetic design principles and wearable fashion collections tailored for individuals seeking balanced and contemporary wardrobe inspiration
Many users browsing digital stores prefer websites that combine speed with simple navigation especially when they access Orchard Merchant Global Shop Pages load quickly and information is presented clearly making browsing smooth intuitive and efficient for users who want easy access to organized product listings online.
Как банки относятся к ипотеке на [url=https://kapsulnyj-dom-1.ru]капсульный дом[/url] — есть ли такая практика?
Online shoppers who appreciate refined digital storefronts often enjoy platforms that combine elegant presentation with a clean structured layout making product browsing more visually pleasing and easy to navigate for everyday purchasing needs Dawn Ridge Atelier Market – offering an elegant atelier inspired shopping experience where products are displayed with clean structure and thoughtful presentation designed to help users browse items smoothly while enjoying a visually balanced and organized online environment
[url=https://geo-optimizaciya-sajta.ru]Гео оптимизация сайта[/url] — поддомены или геостраницы, что лучше выбрать?
During a casual browsing session across various websites, I noticed visit this link – The design is clean and simple, and nothing feels cluttered while moving through sections, which makes navigation easy and comfortable overall.
As I explored different film and cinema platforms, I noticed this Folie a Deux film site and film related content appears artistic, engaging, and thoughtfully presented style, offering a visually creative layout that feels immersive and well designed.
While exploring various online educational resources and creative marketplaces that aim to improve user engagement and discovery experiences across different topics Pure Value Outlet Hub – the platform stands out as an engaging environment that encourages exploration and idea development with a smooth learning flow and consistently inspiring creative tools for users at all levels.
Users exploring online catalogs often value fast navigation and clean layouts when visiting Clever Goods Discovery Arena as it allows them to quickly scan available products and move smoothly between categories without losing focus or encountering delays.
While exploring several online options, I came across visit here now – It feels straightforward overall, and moving through the site was simple and not confusing at all during browsing.
While reviewing ecommerce platforms optimized for cart flexibility and checkout clarity, I noticed that adaptable cart systems improve navigation and usability, which stood out when exploring flexible checkout cart hub – The cart feels easy to adjust, and the checkout flow seems quick, simple, and well structured.
Users looking for well organized product listings often come across pages featuring corner product showcase which enhances clarity and makes browsing simpler while supporting easy comparison of items across multiple categories online for better shopping experience today smoothly across devices everywhere
Consumers exploring e-commerce websites frequently look for platforms that simplify shopping and highlight useful offers where smart buy nest center is included in descriptions and it represents a structured marketplace designed to improve usability while ensuring users can easily access deals and complete purchases without unnecessary complexity or delays in navigation.
услуги поискового продвижения [url=https://34355.ru/novosti/raznoe/5363-0]услуги поискового продвижения[/url]
While browsing online information directories, I came across this OneCentral resource portal and got a good first impression, as the content appears organized and useful, making it easy to understand what the site offers at a glance.
Many individuals browsing online stores appreciate platforms that combine smart technology with user friendly interfaces for smoother transactions smart consumer hub portal enabling easier access to essential items and improved shopping efficiency – It demonstrates how technology integration improves convenience in modern retail platforms.
While going through several entrepreneurship resources, I discovered this informative business link and found the material to be well-structured, providing actionable tips that feel realistic and applicable to everyday business challenges.
In the course of evaluating online shopping systems focused on structured browsing and usability, I found that clean shopping hubs enhance flow and reduce friction, which became evident when exploring smooth retail navigation hub – The platform is well organized, making browsing different sections feel simple and seamless.
In the process of evaluating digital shopping platforms focused on layout clarity and browsing flow, I found that stacked designs enhance product discovery by keeping everything in a smooth scrollable structure, which was clear when reviewing organized stack marketplace hub – The interface feels well structured and easy to navigate, allowing users to browse products effortlessly through stacked sections.
Fashion lovers often explore online brands that present modern clothing collections inspired by aesthetic design and contemporary fashion trends for versatile styling Elegant Apparel Fashion Studio – showcasing a curated platform of stylish clothing and aesthetic focused collections designed to support modern wardrobe needs while emphasizing creativity comfort and refined personal expression
Shoppers frequently appreciate websites that offer clean design and easy navigation especially when they visit Seaside District Frost Goods The platform looks neat and well structured providing a simple browsing experience that feels comfortable and easy to use.
While casually browsing through different platforms, I discovered go to this page – It appears well structured overall, and navigation feels natural and very straightforward, making the experience simple and easy to follow.
Как [url=https://9online.crforum.ru/viewtopic.php?t=35054]Serm[/url] связан с SEO и влияет ли он на позиции сайта?
Individuals who value simplicity in online shopping often choose marketplaces that provide clear navigation and well structured product listings for everyday essentials and needs Practical Merchant Browse Hub – presenting a clean and efficient shopping platform focused on usability and convenience allowing users to explore products and complete transactions in a straightforward and organized digital environment
While conducting comparative research on ecommerce UX and speed-optimized systems, I observed that fast-loading carts improve efficiency and user satisfaction, which was clear when testing quick access cart portal – The shopping corner responds instantly, making navigation and browsing extremely smooth.
While browsing curated wardrobe platforms, I discovered handpicked preloved wardrobe and the items feel thoughtfully selected, offering a sense of personality and sustainability that appeals to modern fashion enthusiasts. – It feels warm, intentional, and visually balanced overall today now experience here.
продажа базы данных [url=https://gobasego-2.ru/]gobasego-2.ru[/url] .
Online retail platforms that prioritize usability and clarity tend to attract more engaged users who value structured browsing and quick access to products Goods Station Explorer – Intuitive design ensures users can move between categories easily while maintaining focus on relevant items and enjoying a smooth overall shopping journey.
When analyzing interactive platforms built for learning and innovation, experts often highlight the importance of seamless navigation and well organized content presentation systems Value Outlet Idea Network – the site provides a comfortable browsing experience where users can easily explore ideas and engage with content that stimulates creative thinking.
Many shoppers prefer tools that reduce decision fatigue, and they often use Quick pick assistant hub which supports fast browsing and helps highlight relevant items based on user interest while maintaining a clean and efficient shopping environment that improves overall experience – designed for quick and simple selection.
During a quick browsing session, I noticed this useful link – It offers a clean and simple structure that helps users quickly understand how the site is organized without unnecessary complexity.
Shoppers searching for reliable online marketplaces usually prefer websites that provide fresh promotions and easy navigation where easy deals update portal is featured in guides and it highlights a system designed to make online shopping easier while ensuring users can efficiently browse products and enjoy a smooth and fast shopping experience overall.
Shoppers who prioritize trust in online purchasing usually prefer platforms that guarantee safety and responsive service where trusted shopping assistance center is featured in guides and it reflects a marketplace designed to ensure secure transactions while providing reliable support and maintaining user satisfaction throughout the entire shopping experience.
While going through positive message pages, I discovered this supportive inspiration site and really like the message behind this, feels genuine and supportive, offering words that feel kind, simple, and emotionally warm.
People exploring online shopping options often appreciate platforms that keep navigation simple and product access direct, especially when considering services that bring organized listings and helpful browsing tools together for a smoother customer experience overall Online Deck Market which presents products in a well-organized manner while helping users find relevant items without unnecessary effort – The service design aims to reduce complexity while improving how users interact with online catalogs
While evaluating ecommerce platforms focused on cart management and streamlined checkout systems, I noticed that functional design greatly improves usability and flow, which became clear when analyzing open cart commerce toolkit – The cart solution appears functional, and the shopping steps are simple, clear, and easy to follow throughout the process.
Fashion oriented users often seek brands that highlight stylish and minimalist clothing collections designed with modern aesthetic influences for everyday outfits Stylish Aesthetic Apparel Hub – offering a curated fashion platform featuring elegant clothing collections and contemporary designs that focus on comfort style and versatility for individuals who appreciate modern wardrobe aesthetics
While conducting usability testing of ecommerce systems with structured navigation design, I found a listing labeled organized port shopping hub – The platform presents a clean and intuitive layout that makes browsing simple, allowing users to navigate categories smoothly and find products without unnecessary complexity or confusion.
During a comparative study of online retail systems emphasizing cart optimization and seamless checkout design, I discovered that structured cart solutions enhance usability and speed, which stood out when analyzing smart shopping cart portal – The platform delivers efficient cart functionality, ensuring browsing and checkout remain smooth and easy throughout the experience.
While checking out multiple online resources, I stumbled upon click here now – It features a pleasant layout and simple flow, making everything easy to understand at first glance during browsing.
заказать кухню в интернете [url=https://zakazat-kuhnyu-18.ru]заказать кухню в интернете[/url]
As I continued browsing scenic retreat websites, I noticed this Fisherman’s Retreat holiday escape page and the location feels peaceful, scenic, and ideal for relaxing stays experience, with visuals that feel calming and naturally beautiful.
сколько стоит заказать кухню по размерам [url=https://zakazat-kuhnyu-17.ru]https://zakazat-kuhnyu-17.ru[/url]
заказать индивидуальную кухню [url=https://zakazat-kuhnyu-19.ru]https://zakazat-kuhnyu-19.ru[/url]
заказать кухню через интернет [url=https://zakazat-kuhnyu-20.ru]заказать кухню через интернет[/url]
While browsing various online shopping platforms, users often prioritize clarity, structure, and ease of movement between categories, especially when they land on modern stores like Clever Goods Zone Hub where the overall interface supports effortless product discovery and makes navigation feel natural, smooth, and surprisingly efficient for everyday shopping tasks.
When analyzing online vendor platforms and their performance in terms of usability and design clarity, many testers note that well-organized sections greatly improve shopping efficiency while browsing through Harbor Hazel Vendor Works – Smooth browsing experience, products are clearly displayed and accessible, helping users stay focused while exploring categories that are logically arranged for easier decision-making and smoother navigation overall experience.
As I was checking out different recommended sites, I discovered open this page – It gives a strong impression of being well maintained, with a neat and cohesive design that feels carefully put together.
While going through different informational campaign pages, I discovered this Simpsons-themed resource page and this looks meaningful and worth spending some time exploring, with content that seems organized in a way that invites further attention.
While conducting usability research on ecommerce systems focused on smart navigation strategies, I noticed that smart hub designs improve user satisfaction by reducing complexity and enhancing product visibility, which became evident when analyzing intelligent shopping flow hub – The smart hub approach makes shopping efficient and structured, allowing users to browse easily through a clean and intuitive interface.
During usability testing of digital shopping systems focused on simplicity and clarity, I discovered that minimal cart structures improve satisfaction and ease of navigation, which became evident when exploring simple order experience hub – The design is clean and straightforward, making shopping easy and free from any confusion.
During my review of online stores, I encountered this product discovery page and found some appealing products, with navigation feeling fast and convenient, making the overall shopping process smooth and easy to handle.
In digital shopping environments users frequently prioritize fast loading and simple layouts especially when visiting Marble Commerce Cove Hub The interface is straightforward and works really well for quick browsing helping users move through sections without confusion or unnecessary delay.
Individuals who prefer organized online marketplaces often appreciate platforms that use merchant lane systems to divide products into clear sections that improve browsing flow and usability Coast Maple Seamless Lane Hub – featuring a structured e commerce platform where products are displayed in a merchant lane layout designed to make browsing intuitive and efficient for all users
Many users browsing e-commerce websites expect intuitive navigation paired with modern visual appeal that enhances usability, particularly when they visit Trend Insight Hub – The interface feels polished and trendy, making it easy for users to browse comfortably while maintaining a smooth and engaging experience across all sections of the platform.
Consumers often look for e commerce platforms that streamline browsing and provide a consistent experience across all sections of the website, and such a service is SmartSpend Store – It focuses on delivering value driven shopping with a clean interface that supports fast navigation and helps users complete transactions efficiently without unnecessary distractions
Across evaluations of marketplace usability and online retail performance optimization strategies HoneyCove Marketplace Usability Grid analysts point out that structured layouts enhance efficiency – users benefit from quick product access and simplified comparison tools during browsing.
In the course of evaluating online shopping systems focused on smart categorization and accessibility, I found that structured marketplaces enhance usability and clarity, which became evident when analyzing smart marketplace navigation portal – The products are well organized, allowing users to find items quickly and easily.
During a comparative study of online marketplaces focused on user experience, I found a section called warm nest browsing center – The layout feels soft and structured, allowing users to explore products comfortably while maintaining a smooth and enjoyable navigation flow across all categories.
In e-commerce environments users often value structured navigation and simplicity especially when accessing Stone Collective Harbor Vendor Network The platform felt smooth overall and there was no confusion while navigating making the browsing experience easy intuitive and enjoyable from start to finish.
As I explored various online platforms, I reached open this resource and found the content neat and organized, with simple navigation that made everything easy to browse through.
People who enjoy creative online shopping platforms often look for websites that use trading post themes to create dynamic browsing environments with varied product selections that make shopping more interesting and enjoyable Wave Harbor Select Goods Hub – providing a clean e commerce platform where trading post design enhances product variety and ensures a dynamic browsing experience that feels smooth and visually engaging across all categories
Many digital users appreciate trend-based platforms that offer fast navigation and organized content presentation, especially when browsing Clever Trends Corner Point – The interface makes discovery simple and enjoyable, helping users move through categories smoothly while maintaining a clean and modern browsing structure across all pages.
In the process of evaluating digital shopping platforms focused on promotions and savings optimization, I found that deal oriented layouts enhance user satisfaction by making discounts more visible, which was evident when reviewing value deal discovery hub – The deals seem attractive and convincing, making it feel like a good destination for users who want to maximize savings during online shopping.
HoneyCoveVendorStudio – Clean interface, everything loads fast and works very smoothly.
In the process of evaluating ecommerce UX systems for clarity and structure, I discovered a section called tree style browsing hub – The interface provides a clean hierarchical layout that allows users to navigate categories easily and find products efficiently while maintaining a smooth and intuitive shopping experience overall.
I was checking different outlet-themed websites for general comparison and usability testing purposes when I encountered a page that stood out for its simplicity Hollow Creek retail index and easy structure which makes it approachable even for first time visitors – It leaves a calm impression with no unnecessary complexity in navigation
While browsing fundraising motorsport sites, I came across charity karting event hub and interesting concept overall, seems well organized and quite engaging today, with a clean design that connects racing events to charitable goals in an understandable way. – The experience feels engaging and structured.
While testing different deal aggregation websites, I discovered organized deals hub which compiles various offers into neat sections – this shopnetmarket.shop service enhances browsing speed, reduces confusion, and allows shoppers to focus on relevant discounts while maintaining a smooth and straightforward user experience throughout their online shopping journey.
During a routine browsing session, I found check it out and noticed the content was cleanly structured with easy navigation, making the overall experience smooth and fairly enjoyable.
While analyzing ecommerce UX designs centered on global cart systems and international shopping convenience, I observed that unified carts improve usability and satisfaction, which was evident when reviewing global shopping experience portal – The design reflects a global cart style, offering a wide range of online products in a structured layout.
Online buyers benefit from platforms that consolidate multiple deals into one easy to navigate shopping environment ShopEdge Connect Zone – The system integrates various promotions into a unified interface for easier shopping decisions.
People who appreciate calm digital shopping platforms often look for websites that combine lakefront themes with lounge style layouts to make browsing feel soothing and visually engaging Lakefront Velvet Flow Market Hub – providing a clean shopping environment where lakefront inspired visuals improve product display and create a smooth browsing experience designed to help users explore items comfortably and efficiently
In the course of checking various online shopping directories and vendor ecosystems, I arrived at Local vendors collective browsing link which appeared to streamline access to multiple vendors and product categories making it easier to explore different shops within a single platform – overall it felt organized and accessible for browsing purposes
Online users often prefer e-commerce platforms that maintain a balance between simplicity and functionality, particularly on CleverMart Smart Hub – Everything is well organized, allowing users to browse comfortably while enjoying a smooth, structured, and visually consistent shopping experience across the site.
While reviewing digital shopping directories, I discovered fast browse corner hub which structures listings into easy categories – Hyper Cart Corner provides a really easy shopping experience with affordable products and fast delivery, helping users save time while searching for deals and enjoying a more organized browsing experience overall.
When examining online retail platforms built for enhanced usability and optimized browsing workflows across diverse digital product ecosystems and vendor listings Trading Foundry Icicle Link feedback indicates high satisfaction – Overall experience remains simple and effective with fast navigation clear interface structure and well organized product displays supporting effortless comparison and exploration of available items.
Many shoppers prefer online platforms that function as neon shopping centers with helpful product ranges and intuitive navigation for seamless browsing Neon Cart Ease Hub Point improving usability – It is commonly appreciated for its simple design and well organized product listings
Online shoppers frequently value websites that act as premium goods corners with solid collections of items and smooth browsing experiences for quick purchasing Premium Goods Vision Corner Flow making shopping easier – It is commonly noted for its clean layout and smooth checkout system
While evaluating different shopping hub websites, I came across a resource that seemed well structured and easy to understand Hollow Creek discovery page with a layout that supports straightforward navigation – It gives a pleasant browsing experience without unnecessary complexity at first glance overall
While exploring digital marketplaces for better deal tracking, I found streamlined offers tracker that organizes promotions effectively – this shopnetmarket.shop system supports smoother navigation, allowing users to locate relevant discounts faster and improve their overall shopping experience through simplified categorization and clear product presentation.
In e-commerce browsing experiences users often expect clarity and structured design especially when accessing Cove Goods Forest Atelier The layout is clean and tidy giving a professional impression that allows users to navigate easily and explore products without confusion or clutter.
Many shoppers enjoy rapid goods zones offering fast shopping zone with simple layout and useful product range, helping them browse efficiently while maintaining clarity and ease of use FlowSelect Rapid Goods Zone Access – Designed for smooth transitions and fast category switching
While conducting usability research on ecommerce systems emphasizing modern cart design and simplicity, I noticed that corner-based layouts improve browsing comfort and flow, which became evident when analyzing sleek shopping cart portal – The interface feels clean and modern, offering a user friendly experience that makes shopping straightforward and enjoyable.
Online buyers often look for clean interfaces with easy product discovery tools UX Shopping Arena – A user friendly design approach enhances engagement by making navigation straightforward and ensuring that product categories are easy to explore without unnecessary complications or confusion
While exploring various digital commerce ecosystems and marketplace directories, I came across Ember Meadow retail guild index which seems to consolidate shop listings in a unified layout, and after browsing I noticed clear formatting and responsive navigation – overall it appeared clean and efficient
Many people searching for savings prefer digital platforms that regularly update their listings with current discounts and promotions Hot deals listing ensuring that users always have access to fresh opportunities for reducing costs on everyday purchases – A practical solution for staying updated with ongoing sales and limited time offers
Many digital shoppers prefer online stores that provide simple navigation and fast response times, especially when they access Crystal Hub Shopping Point – The browsing experience remains easy and dependable, allowing users to explore categories without unnecessary complexity or confusion during their session.
Across usability evaluations of digital retail environments, consistency in interface design and navigation clarity is repeatedly highlighted as essential for positive user experience Icicle Commerce Bazaar Hub the platform offers an intuitive browsing flow where products are well arranged and easily accessible, making shopping efficient and straightforward for all users.
During a casual online search session, I stumbled upon check this site and noticed that the content looked fairly informative at first glance, so I may return later to explore everything more thoroughly when I get the chance.
Online consumers frequently enjoy platforms that combine curated marketplace picks with fast browsing tools for better convenience and usability Neon Pick Glow Hub Access enhancing browsing – The website is often recognized for its clean layout and smooth category navigation
When trying to find the best online promotions, having a dedicated reference point can be useful, and I noticed this resource included in discussions offer search assistant – it emphasizes helping users locate relevant deals faster while maintaining a simple and accessible browsing experience for all kinds of shoppers.
Users browsing modern e-commerce platforms often value clarity and usability especially on Commerce Atelier Forest Cove The layout is simple and intuitive making everything easy to find while ensuring a smooth browsing experience where users can quickly access different sections without confusion or unnecessary effort.
Users often prefer streamlined fashion spaces like rapid style corner platforms offering stylish items available here with quick browsing and smooth interface, ensuring a more enjoyable experience when searching for trendy products across multiple sections StyleFlow Rapid Corner Trend Center – Designed for clarity and speed, it enhances browsing efficiency and supports smooth product discovery
During a comparative study of online retail systems focused on product diversity and centralized shopping, I discovered that all-in-one stores enhance usability by reducing the need to switch platforms, which stood out when analyzing complete product variety portal – The store offers a broad selection of items, making it feel like a convenient place to find everything in one location.
During an evaluation of different marketplace and outlet-style websites, I found a platform that stood out for its minimal design approach, organized structure, and smooth navigation flow that helps users browse efficiently even during longer browsing sessions online Hollow Ridge product showcase which feels clean and logically arranged for browsing ease – The interface maintains a calm visual structure that makes it easy to focus on content without unnecessary visual noise or complexity.
https://justpaste.it/aca-203
ACA 203 touches down in Tashkent with a flyweight showdown with big implications and a card that leans heavily into the promotion’s local dominance across Uzbekistan.
Many online shoppers value structured browsing experiences that reduce confusion while exploring large catalogs through intuitive interfaces where Shop Central Hub – central browsing concept improves navigation efficiency and helps users locate products faster across categories with smoother transitions and clearer layout organization overall
Many users prefer e-commerce environments that combine multiple product ranges into one organized space, reducing the need to switch between different websites or platforms MegaRange Shopping Center – This approach helps customers access a broad selection of items in one place while ensuring clarity, structure, and improved shopping efficiency throughout the entire browsing process.
People exploring online stores often seek platforms that help them compare products easily while offering a clean and distraction free browsing environment Rapid Finds Store – It is generally viewed as a helpful option for users who prefer quick access to various categories without complicated menu structures slowing them down
During research into independent online storefront listings and vendor aggregation sites I encountered a listing pointing to Ivory Cove commerce showcase index which was embedded in a structured directory and after briefly browsing it I found the content clearly arranged – overall it seemed like a useful platform and I enjoyed moving through its sections smoothly
For those seeking a fight card where the odds-setters more or less checked the stats and left it at that , ACA 203 taking place in Tashkent is a treasure trove you won’t want to miss.
Across studies of modern online marketplaces emphasizing clarity and structured browsing experiences Icicle Isle Vendor Hub – Extensive selection of items is displayed in an organized format, making it easier for users to explore categories and compare offerings efficiently.
https://differ.blog/p/thunder-in-tashkent-the-high-stakes-of-aca-203-9150d2
ACA 203 isn’t just another stop on the regional circuit ; instead, it serves as a career crossroads .
Users browsing e-commerce platforms often expect well organized pages and fast loading speed especially when visiting Digital Buy Central Arena – The site provides a user friendly layout that helps shoppers move easily between sections while maintaining smooth and reliable performance throughout.
Online consumers frequently enjoy marketplaces that combine trendy product offerings with a simple interface design for a more seamless browsing experience Neon Style Bazaar Explorer Hub making browsing easier – The website is often noted for its modern layout and clear product organization that simplifies shopping
Many users browsing digital stores value refined presentation and usability especially when they visit Jasper Trading Gallery Harbor Center After a quick look it feels polished and thoughtfully designed making navigation smooth and easy while maintaining a clean and professional visual style.
During analysis of online retail systems focused on layout consistency and usability, I discovered that stacked product arrangements improve browsing efficiency, which became clear when testing smart stack product center – The layout is well structured and easy to follow, making browsing feel organized and simple overall.
In my review of online retail tools focused on simplicity, I came across minimal cart system that organizes shopping categories neatly – Simple shopping experience with clear layout and easy product access ensures users can quickly browse products and make decisions without unnecessary complexity or excessive steps during the shopping process.
Users often prefer structured e-commerce environments like rapid trend hubs offering trend hub offering fresh items and easy navigation for users, ensuring a simplified shopping journey that reduces effort while improving product discovery efficiency across multiple sections TrendFlow Rapid Hub Shopping Center – Designed for speed and usability, it enhances navigation flow and provides organized category browsing
Shoppers increasingly prefer organized e-commerce layouts that reduce friction during browsing CartFlow Market Center provides structured category access while ensuring users can move between sections smoothly with improved usability and a more intuitive shopping experience designed for everyday customers overall experience.
While exploring various online retail environments and comparing their structural design approaches, I came across a store that offered a minimal yet functional layout that made browsing feel efficient and easy to manage even for first-time visitors navigating through categories Honey Fern digital store providing a structured entry point for product exploration and category viewing – The overall experience feels clean, balanced, and designed to support effortless browsing without unnecessary distractions.
Shoppers who value practicality often choose platforms that allow them to browse easily and complete transactions without dealing with unnecessary steps or confusing layouts Practical Items Hub – It provides a clean and efficient shopping flow that supports quick product selection and a straightforward checkout process for improved user satisfaction
While exploring independent marketplace systems and digital shop directories I encountered a listing pointing to Ivory Ridge commerce listing index which appeared in a curated directory and after a short look I noticed the layout was minimal and readable – overall the browsing experience felt organized and simple with smooth navigation throughout
During a short browsing session earlier, I came across see more details and found the website interesting overall, with a few useful bits that I discovered while scrolling casually through the content.
While going through several pages online to compare designs, I discovered explore this content here – The structure feels organized, making navigation simple and comfortable for users.
IvoryBrookVendorFoundry – Clean design, shopping feels smooth and very easy today.
While exploring modern online shopping platforms users often look for clarity speed and structured layouts that improve browsing efficiency especially when they visit Digital Cart Arena Hub – Cart browsing feels smooth and products are displayed in a clear and organized way that makes it easy for users to find items without confusion or delays.
While analyzing ecommerce UX designs centered on innovation and digital shopping trends, I observed that tech-focused systems improve navigation and engagement, which was evident when reviewing modern tech browsing center – The platform feels contemporary and appealing, offering products that align with modern user interests.
Online consumers frequently prefer platforms that gather modern trends and updated product selections in one central place so they can shop efficiently every day Neon Style Trend Explorer Hub making browsing smoother – The website is often noted for its structured design and visually appealing layout that simplifies shopping
Online users often prefer e-commerce websites that ensure fast transitions and clean layouts especially when they access Floraridge Vendor Craft Atelier The platform offers smooth browsing where pages load and switch quickly without lag improving overall usability and user satisfaction.
Many users trust rapid trend outlets that feature outlet deals on trending products with simple and clean structure, ensuring a smooth and enjoyable shopping journey across all product sections ClearView Rapid Trend Outlet Center – Focuses on structured layout and fast product access
E-commerce platforms are valued most when they provide seamless navigation and dependable checkout experiences overall in modern shopping today Direct Order Hub optimized for speed – the system helps users move through categories efficiently while maintaining a consistent and reliable interface
Online shoppers often seek platforms that deliver fast browsing performance combined with structured product displays and intuitive navigation tools CartStyle Navigation Zone – this design enhances usability by ensuring clear product organization and enabling users to move seamlessly between different shopping categories overall.
Many online buyers prefer platforms that simplify product searching while still offering a wide selection of useful items across different categories and needs Smart Shopping Route enabling them to complete purchases quickly without dealing with complicated menus or confusing layouts – The site is generally seen as efficient and well structured with a focus on user convenience
E-commerce platforms are rapidly evolving to meet user expectations for speed, personalization, and easy access to diverse product selections on global scale today Digital Tide Store illustrates how modern online systems adapt to changing consumer behavior – Wave market brings streamlined browsing tools that improve accessibility and enhance overall user satisfaction in digital shopping
During my search through independent vendor directories and online shopping networks I discovered Ember Brook collective storefront index which compiles various shop listings and product categories into a single browsing interface – I considered it moderately useful for exploring niche sellers
While scanning digital storefront designs, I found a website that offered a clean and minimal browsing experience, helping users move through sections efficiently with integrated elements like a href=”[https://indigoharborstore.shop/](https://indigoharborstore.shop/)” />Indigo Harbor marketplace link placed within the page structure – The overall feel is intuitive and user-friendly, with a strong emphasis on easy navigation and content clarity.
Across various marketplace reviews performance and usability are frequently tied to how well platforms structure their product categories and navigation flow Cove Ivory Commerce Link browsing remains straightforward with clean design and responsive layout across pages improving overall usability today
While reviewing ecommerce UX systems optimized for continuous browsing experiences, I observed that flow-based shopping models reduce navigation barriers and improve clarity, which became clear when testing seamless shopping flow center – The design ensures smooth navigation throughout sections, helping users move easily between products and categories.
As I explored several online pages out of curiosity, I landed on explore here and gave it a quick look, finding the design clean and quite user friendly, which made the browsing experience feel simple and comfortable.
Many users appreciate platforms that feature a neon inspired trend zone with fresh items and an easy browsing layout for simple shopping Neon Trend Market Zone Hub making browsing smoother – It is frequently described as efficient and visually appealing with structured categories
Many digital shoppers appreciate e-commerce sites that offer organized cart systems and intuitive design flow especially when visiting Digital Center Cart Shop – The platform helps users navigate cart options effortlessly while maintaining a clean and user friendly interface throughout all browsing sessions.
Many shoppers prefer platforms that emphasize readable content and clean structure especially when they visit Cove Hazel Commerce Studio The content is nicely displayed making it comfortable to read through and ensuring a smooth and enjoyable browsing experience across all sections.
Users increasingly choose platforms that deliver fast updates and organized layouts to simplify online shopping experiences effectively across modern devices Trend Pulse Center Flow Portal – allowing them to stay informed and browse efficiently without unnecessary distractions or delays during their purchase journey in real time
In the process of exploring ecommerce marketplaces, I found a platform that maintained a structured layout with clear navigation and varied product listings BuyZone retail browsing portal embedded naturally within the page – The marketplace feels well organized and easy to use, offering a good range of products and a seamless browsing experience.
While going through different online pages, I discovered explore this site – The simple design works well, with fast loading pages and a clean visual style.
Pretty section of content. I just stumbled upon your web site and in accession capital to assert that I get actually enjoyed
account your blog posts. Any way I’ll be subscribing to your feeds and even I achievement you
get right of entry to constantly fast.
кухни на заказ [url=https://kuhni-spb-58.ru]кухни на заказ[/url]
During exploration of digital storefront systems and curated commerce networks I found Honey Cove vendor access portal which presented listings in a clean hierarchical layout – overall the structure made navigation easy and the information straightforward to follow while casually browsing
кухня на заказ спб [url=https://kuhni-spb-57.ru]кухня на заказ спб[/url]
While reviewing several online marketplace templates I found one interface that offered a balanced structure and minimal design approach Meadow Ink landing Meadow Ink landing It allows smooth transitions between sections and keeps content organized so users can browse without distraction or unnecessary complexity appearing anywhere across the page layout consistently overall experience flow
ленинградские кухни [url=https://kuhni-spb-59.ru]https://kuhni-spb-59.ru[/url]
Across various online marketplace comparisons, structured design and simple navigation are consistently linked to improved user experience, and within this platform Ridge Vendor Commerce Hub the system maintains a clear and accessible layout that helps users browse easily and understand product categories without difficulty.
m fortune https://m-fortune.info/
People exploring digital marketplaces often appreciate reliable cart stations with smooth checkout and organized shopping flow for seamless transactions Next Cart Flow Bright Hub making browsing smoother – It is commonly described as modern and user friendly with structured purchasing tools
Online users often prefer e-commerce websites that maintain a clean and simple structure especially when they access Icicle Foundry Trading Brook The simplicity is noticeable and nothing feels cluttered which makes browsing smooth comfortable and easy to follow from page to page.
Online shoppers often seek refined cart systems that streamline purchasing steps and offer visually organized interfaces for easier decision making during browsing Cart Royal Navigator Experience Center – built to support efficient shopping journeys with smooth checkout flow, structured product pages, and simplified navigation for all users
Online users often prefer shopping platforms that combine simplicity and speed in cart management especially when they visit Cart Zone Easy Shop – The interface is well structured and improves usability creating a smooth and efficient shopping experience across all product categories.
Users looking for smarter shopping experiences often gravitate toward websites that simplify essential product selection and reduce time spent searching through categories smart essentials market – This kind of service is appreciated for offering a structured environment where users can easily find what they need without unnecessary complexity or confusion
Online buyers generally prefer platforms that combine wide product selection with easy browsing tools for a better shopping experience overall Universal Cart Navigator helping users shop with confidence and ease – The website is known for its organized structure and smooth browsing functionality
Style saver markets attract users who want to maintain a fashionable lifestyle while also taking advantage of discounts and promotional offers available online Style Saver Market It delivers a mix of trendy products and ongoing savings enabling users to shop smartly while enjoying a wide range of stylish and affordable options regularly
While scanning niche marketplace aggregators and digital vendor directories I came across studio vendor network portal which appeared within a structured commerce listing and after checking it briefly I found the layout readable – overall it seemed like a decent platform and the navigation experience was straightforward and uncomplicated
Online customers who prefer well arranged shopping environments can explore platforms featuring Modern Cart Marketplace within their system, delivering categorized product listings, smooth navigation, and an efficient browsing structure that helps users quickly compare options and enjoy a more comfortable and productive digital shopping experience from start to finish.
While reviewing ecommerce UX systems designed for improved cart flow and usability, I observed that smart cart corner designs reduce friction and improve transaction speed, which became clear when testing simple checkout cart index – The cart corner layout feels practical, with a clean and simple checkout flow that makes purchasing easy and stress free for users.
While browsing through different online options, I came across view this bold page – The straightforward layout makes it easy to find what you need with minimal effort.
кухни на заказ в спб от производителя [url=https://kuhni-spb-61.ru]кухни на заказ в спб от производителя[/url]
кухни на заказ в спб недорого [url=https://kuhni-spb-60.ru]кухни на заказ в спб недорого[/url]
During a review of modern boutique style online stores I encountered a website that focused on clarity organization and minimal interface design Iron Hollow curated boutique portal making it ideal for users who prefer straightforward navigation and a calm browsing experience while exploring fashion related products online
When assessing digital marketplace efficiency and responsiveness in handling large-scale product listings and user interactions, Brook Jasper Trade Nexus is recognized as fast loading pages contribute to a smooth shopping experience that feels dependable and consistent for users exploring items across multiple categories.
Online shoppers appreciate systems that present discounts clearly while maintaining a refined and elegant design for better usability and faster decision making Deal Harmony Royal Experience Hub – focusing on structured navigation, clear promotional visibility, and efficient browsing flow that enhances the overall online shopping experience significantly for users
Online buyers often value websites that function as next generation buy hubs with smart deals and diverse product selections for better convenience Next Gen Cart Buy Hub Flow making shopping smoother – It is commonly described as user friendly with structured categories and simple checkout processes
Users exploring online marketplaces frequently appreciate structured layouts and fast performance especially when visiting Orchard Commerce Drift Hub Pages load quickly and information is displayed clearly making browsing smooth intuitive and easy for users who want efficient access to organized product content across the site.
E-commerce systems today focus heavily on delivering seamless experiences that reduce friction during online shopping Seamless Shop Entry – The entry point to the platform is optimized for ease, helping users start shopping without delays or confusion right from beginning immediately
In modern e-commerce environments users frequently prioritize usability and clear design flow especially when visiting Trend Corner Smart Hub – The trend section is structured in a way that makes browsing smooth and efficient while providing a clean and enjoyable user experience overall.
Many users appreciate platforms that make it easier to discover seasonal promotions without needing to browse multiple separate websites Discount Discovery Hub improving shopping convenience and saving time overall – The site is known for its clean design and well organized promotional categories
Deal enthusiasts enjoy platforms that bring together a wide range of discounts in one accessible place allowing them to explore options without switching between multiple websites Elite Discount Zoneboard – This variation presents a more premium styled zone where exclusive deals are organized in a structured board format helping users quickly identify high value opportunities with minimal browsing effort
While browsing through independent retail platforms and curated online shop collections I discovered Ember Cove shop network index page which compiles multiple vendor listings into a structured browsing environment and the experience felt smooth with fast loading times and content that is easy to read – it gave a decent overall usability impression
During analysis of structured ecommerce layouts, I came across a section labeled axis retail browsing hub – The design organizes products neatly in a logical flow, making it easy for users to explore items without confusion or unnecessary complexity in the interface design.
I was exploring different online shopping options when I encountered have a look here – The layout is simple yet effective, and the product pricing feels balanced, which makes it look like a comfortable place to browse without pressure.
During a light browsing session online, I visited click to view and found it to be quite helpful, with a pleasant and simple interface that made navigation easy and comfortable.
Users frequently prefer well designed marketplaces that allow smooth exploration of quality goods with clear categories and fast navigation systems Royal Goods Experience Flow Arena – offering simplified browsing structure, efficient product discovery tools, and a user friendly interface that enhances online shopping satisfaction and clarity
In the process of analyzing ecommerce marketplace structures I found a platform that emphasized clarity and structured presentation Leaf Iron shop overview included within the content flow and making navigation feel simple and intuitive – The design maintains a balanced layout that helps users understand categories easily and browse through sections without confusion
When analyzing user experience trends in e commerce platforms, researchers often emphasize how consistent design patterns and structured layouts contribute to better engagement and smoother browsing journeys across all device types and screen sizes Jasper Cove Retail Flow – The platform feels reliable and easy to use, with well arranged sections that support quick navigation and efficient browsing.
Shoppers who value efficiency in digital marketplaces often choose platforms that make browsing and checkout feel effortless and fast Instant Cart Access Hub enabling users to move through categories without confusion while maintaining a responsive and clean interface overall – This setup emphasizes clarity and speed so users can complete purchases without unnecessary interruptions or complexity
Users exploring modern digital shops frequently prefer websites that combine clarity and performance especially when visiting Fern Cove Market Atelier It looks reliable and well structured offering a browsing experience that feels smooth organized and comfortable for users navigating through product selections.
https://ecopolis-spb.ru/tvorchestvo_all/
Shoppers today often look for digital marketplaces that simplify product discovery by organizing items into clear and easy to navigate sections Smart Goods Browser Hub making online shopping more efficient and less time consuming – The platform is recognized for its straightforward interface and smooth category transitions
Many shoppers prefer digital marketplaces that offer clear navigation paths and structured deal sections especially when they explore Digital Deal Smart Zone – The browsing experience remains smooth and efficient with quick access to deals that makes shopping easy and enjoyable throughout.
pravdadaily.com.ua/elfiyski-imena-naykrasyvishi-ta-naypopuliarnishi-varianty-dlia-personazhiv/
Users looking for comprehensive shopping experiences often value platforms that bring together many categories in one structured and easy to navigate place Ultimate Goods Style Arena – It is presented as an all in one shopping arena where stylish products are grouped efficiently allowing users to explore a wide variety of goods seamlessly
During review of curated online retail hubs and independent seller listings I encountered Flora Brook vendor showcase index which organized content into simple categories with a clean browsing interface – I noticed some useful insights while casually going through the marketplace pages
E-commerce platforms focus heavily on building trust through consistent service quality and accurate product information Accurate Trust Shopping Network – It delivers reliable listings and transparent information that help users make informed and confident purchasing decisions.
During a quick search for new shopping ideas, I encountered view this page now – The layout feels simple and smooth, and the items look appealing enough to make browsing feel easy and enjoyable.
During a comparative study of online retail interfaces emphasizing navigation clarity and flow systems, I discovered that route-style layouts improve usability by guiding users step by step through product categories, which stood out when exploring guided route marketplace portal – The navigation system helps users move through sections easily, making product discovery feel intuitive and well structured without confusion or unnecessary complexity.
Many online buyers prefer stations that highlight updated products in a clear layout, helping them browse faster and make better shopping decisions overall Royal Goods Smart Navigation Station – offering organized browsing flow, fast access to categories, and a simplified shopping interface designed for improved user satisfaction and usability
When looking into scalable solutions for online retail platforms, I came across web shop manager that offers structured tools for better store organization – it improves operational flow, enhances usability for administrators, and supports the creation of flexible e-commerce environments that can grow alongside business needs without unnecessary technical limitations or complications.
Many shoppers prefer online stores that provide clean layouts and easy usability especially when they land on Fern Vendor Corner Hub The interface is straightforward and I found it easy to navigate and locate essential content without confusion or unnecessary effort.
When reviewing online shopping environments focused on accessibility and performance, experts frequently mention the importance of clean interfaces and easy-to-understand navigation flows Harbor Commerce Visual Depot – Everything is well structured and visually clear, supporting smooth browsing experiences.
Online consumers frequently seek platforms that maintain organized product catalogs and steady updates allowing them to browse categories without difficulty or confusion Goods Update Navigator Hub improving usability – The site is recognized for its clear structure and consistent product listing updates that improve overall shopping experience
During exploration of various ecommerce websites, I came across a platform that delivered strong performance and a visually appealing interface design Petal Iron storefront hub integrated into the content flow – The pages load quickly and the structure remains organized, resulting in a smooth and enjoyable browsing journey.
Online customers looking for premium styled platforms often value both performance and structured organization in product browsing especially when searching for curated selections across multiple categories Prime Stylish Goods Portal Center – This portal center delivers prime stylish goods with a focus on fast navigation and well structured categories designed to improve shopping clarity and speed
Shoppers frequently appreciate online stores that offer intuitive interfaces and fast performance especially when they visit Goods Digital Corner Market – Product listings are well structured and easy to browse making the shopping experience smooth efficient and enjoyable across all categories.
During research into online vendor directories and curated retail platforms I found Flora Brook digital shop index which offered a clean layout with clearly separated marketplace sections – I came across useful insights while casually browsing through its content structure
Shoppers often look for well organized digital stores that allow fast browsing and simple navigation across multiple categories Premium Goods Station Category Flow Hub – The website structure supports efficient browsing and helps users locate desired items without difficulty quickly easily
рулонные шторы автоматические купить [url=https://rulonnye-shtory-s-elektroprivodom17.ru]рулонные шторы автоматические купить[/url]
During a quick search for new online stores, I encountered see this collection now – It looks like a promising platform, offering a variety of products with prices that feel quite reasonable.
During a routine browsing session, I checked out check it out and found the site quite nice overall, with information presented clearly and smooth loading performance that made navigation simple.
Are you looking for a fast, safe and hassle-free transfer in Valencia? We offer professional transport and moving services, both for individuals and companies. Request your budget without obligation and enjoy a quality service, https://trasladoavalencia.es/
https://capital360.com.ua/
Many digital shoppers prefer platforms that keep trend browsing simple and visually appealing, helping them discover products without unnecessary distractions Royal Trend Clean Navigation Portal – offering fast loading pages, structured layouts, and efficient browsing flow that enhances usability and improves overall shopping comfort significantly
Frequent online shoppers value websites that prioritize fast loading pages and easy access to ongoing promotions across multiple product categories and brands Flash Discount Hub – Designed for rapid deal discovery, it ensures users can catch limited time offers immediately while maintaining a smooth and uninterrupted browsing experience across all devices and connection speeds.
People browsing online stores often value websites that showcase trending goods in a visually appealing and well organized layout Infinity Fashion Trend Zone improving flow – It is commonly recognized for its stylish interface and smooth navigation that supports effortless browsing across categories
In e-commerce environments users often expect clean design and comfortable spacing especially when visiting Trading Foundry Brook Forest The layout is nicely spaced out making browsing easy and pleasant while ensuring users can navigate without visual strain or confusion.
Across multiple e-commerce performance reviews, specialists often focus on interface clarity and fast navigation as key drivers of user satisfaction and engagement Jewel Commerce Atelier Flow – Browsing feels easy and comfortable, with a well organized layout that supports smooth exploration of products across categories.
Online shoppers often look for well organized spaces that make browsing easier and more enjoyable across categories Elegant Style Corner this corner provides a smooth experience with neatly arranged product sections and simple navigation allowing users to find stylish items quickly without confusion or unnecessary complexity
While reviewing different websites during a short search, I stopped at learn more stone site and had a quick look where everything appeared neat and easy today, giving a structured and easy browsing experience.
While reviewing online outlet store layouts, I discovered a site that balanced visual appeal with functional usability in a very effective way IronPetal outlet clearance hub embedded within the page design – The overall structure feels smooth and accessible, providing users with an easy browsing experience that supports quick category navigation and clear product visibility.
Modern retail environments are designed to provide streamlined experiences that help users shop efficiently while minimizing unnecessary steps in the purchasing journey SimplePath Cart Hub – The platform offers a direct and uncomplicated shopping flow that improves user satisfaction by focusing on clarity and ease of use
Online users often prefer e-commerce websites that reduce complexity and improve clarity especially when accessing Digital Pick Smart Market – The browsing experience is structured and easy to follow making item selection quick and effortless for all types of shoppers.
Should you be on the hunt for a fight card where the oddsmakers basically just looked at records and called it a day , ACA 203 in Tashkent is a gold mine for you .
In the course of reviewing ecommerce deal directories, I came across a module titled discount savings explorer hub – The interface is simple and well structured, showcasing appealing offers that encourage users to continue browsing and discover more products with competitive and interesting pricing options overall.
During my search for interesting products, I stumbled upon check this out now – The design looks simple and effective, making it easy to move across different categories without confusion or delay.
рулонные шторы на пластиковые окна с электроприводом [url=https://avtomaticheskie-rulonnye-shtory50.ru]рулонные шторы на пластиковые окна с электроприводом[/url]
Shoppers looking for convenient digital marketplaces often value curated selections that help them discover useful products quickly without feeling overwhelmed by too many options Premium Pick Market Smart Browse Hub – The platform stands out for its clean design and well organized structure that enhances user satisfaction and browsing speed
This May, the tension within Tashkent’s Humo Arena won’t just be thick with the roar of a partisan crowd ; it will instead weigh down on the professional lifeline of athletes who have dedicated lifetimes to chasing the ultimate prize.
ACA 203 is far from merely another date on the regional circuit ; it stands as a defining crossroads .
рулонные жалюзи на пластиковые окна купить [url=https://rulonnye-shtory-s-elektroprivodom11.ru]https://rulonnye-shtory-s-elektroprivodom11.ru[/url]
https://telegra.ph/Analyzing-the-Value-at-UAE-Warriors-70-Medkouri-vs-Neves-05-01
UAE Warriors 70, which happens on May 8, 2026, at the Space42 Arena in Abu Dhabi is a prime illustration of this.
Many online users look for trend hubs that mix royal inspired styles with modern product updates, ensuring a balanced and enjoyable browsing environment Royal Trend Smart Browse Center – offering organized categories, quick navigation tools, and a user friendly interface that enhances product discovery and daily shopping experience quality
рулонные шторы электрические [url=https://rulonnye-elektroshtory.ru]рулонные шторы электрические[/url]
I was casually scrolling through a few options online when I came across ambercrest hub and it immediately caught my attention with its simple layout and calm presentation – It really feels like a pleasant place to explore without any rush or confusion at all.
As I moved through a few online pages earlier, I discovered visit hollow shop and it felt very balanced – Browsing here was smooth and easy, and the products look nicely arranged overall, making the site pleasant to explore.
In reviewing e-commerce browsing solutions and product discovery tools, I encountered retail branch market hub which organizes items into logical shopping groups – Shop tree market provides structured categories for shoppers and enhances usability by allowing customers to quickly filter and identify relevant products without unnecessary browsing difficulties or delays.
Online consumers often enjoy websites that display new fashion trends quickly so they can browse and shop without missing recent updates Instant Trend Style Portal making shopping smoother – It is appreciated for its fast content updates and well organized product categories
Online users often value websites that provide clear navigation and strong structure especially when they land on Guild Orchard Granite Hub The platform feels well put together and the presentation is solid giving users a comfortable and efficient browsing experience.
Shoppers exploring digital stores frequently enjoy platforms that offer a sense of discovery while browsing through different product categories and options, particularly on copper petal selection site – items felt interesting and made the experience worthwhile, encouraging another visit to see what new products might appear in the future.
As I browsed through several pages today, I came across surf trading page and found the presentation neat and minimal; I left a quick comment – overall it delivered a smooth experience with clearly arranged information that was easy to read.
Many users exploring online marketplaces appreciate systems that combine speed with clarity especially when browsing multiple product categories in a single session Goods Hub Express – The platform emphasizes fast browsing performance and a hub style organization that allows shoppers to quickly locate items and complete purchases without unnecessary delays
During a random browsing session, I discovered check out this page – The design seems well thought out, and everything loads quickly, helping create a smooth and efficient browsing experience overall.
In e-commerce browsing experiences users often prioritize clarity and intuitive navigation especially when accessing Digital Smart Trend Corner – The trend section is well arranged making browsing smooth and efficient while ensuring users can explore content without unnecessary complexity or delay.
While passing time online and opening different pages, I reached visit this webpage and appreciated how the content felt current and easy to read, creating a pleasant browsing experience overall.
Many digital users appreciate platforms that function as premium pick zones because they provide a nice variety of picks along with fast loading product pages that make browsing smoother and more enjoyable Premium Pick Zone Flow Navigator – The website is known for its structured interface and responsive design that ensures quick transitions between categories and improves overall user satisfaction
Many users enjoy platforms that showcase premium trends in a simple station layout, making browsing faster and more intuitive for daily shopping Smart Elegant Trend Station Access Hub – focused on clean design, smooth navigation, and structured product presentation that improves user experience and shopping efficiency overall
Users who enjoy thematic online stores often find value in platforms that blend natural inspiration with curated product displays, making browsing feel calm, immersive, and thoughtfully paced across different categories and collections echo hollow artisan portal – The concept suggests a rustic digital environment where handcrafted and nature-inspired items are showcased in a way that encourages slow, enjoyable discovery.
Digital shoppers often highlight that engaging product selections make browsing more enjoyable and increase the likelihood of returning to a site, as seen on petal copper goods hub – the experience felt worthwhile since items looked appealing and made the platform feel like a place worth revisiting.
People exploring trendy household product platforms frequently enjoy websites that highlight both innovation and simplicity in how items are presented and described Trendy Home Goods – it showcases stylish and functional products in a way that helps users quickly understand how each item enhances modern living spaces effectively
Online users exploring luxury marketplaces often prefer platforms that highlight exclusive items in a simple and organized format Luxury Goods Discovery Point enhancing experience – The platform is known for its elegant structure and smooth browsing functionality
While exploring different online stores, I came across see hollow items and it gave a soft, neat impression – The browsing experience felt smooth and relaxed, with products nicely arranged in a clear and visually simple way.
Visitors testing out new e commerce platforms usually notice how organization and clarity affect their impression of reliability and usefulness in everyday browsing situations urban corner flash hub the structure feels light and fresh which helps users move through pages without distraction or unnecessary complexity slowing them down
During research into online commerce gallery networks and digital storefront exhibits I came across a structured entry featuring Moon Cove gallery discovery portal which appeared within a curated directory and after reviewing it briefly I noticed the layout was minimal and easy to follow – overall it felt engaging and I would likely return again for future updates
Shoppers who enjoy visually appealing layouts often prefer platforms that combine design with functionality for better engagement Visual Style Zone – The interface blends attractive visuals with easy navigation helping users browse products comfortably and efficiently
During a usability review of modern shopping cart systems, I found a section labeled quick checkout flow portal – The platform is designed for speed, providing instant cart updates and smooth navigation that enhances the overall shopping experience with minimal effort required from users.
Consumers today expect e-commerce systems to deliver personalized recommendations and fast access to relevant deals across a wide range of product categories Smart Deal Junction – The junction connects users to curated offers and smart deal selections designed to enhance shopping efficiency and reduce decision-making time
For casual online shoppers seeking variety, GoldenBay retail showcase platform delivers a structured browsing experience with clearly arranged sections, allowing users to move through categories easily while enjoying an overall seamless and comfortable interface.
ironwavecollective.shop – Collective platform looks solid, easy access and well structured pages
I was exploring various online marketplaces when I discovered explore this page now – The platform appears reliable, and the product listings seem clear and informative, making the browsing experience simple and effective.
I was casually surfing the web when I discovered shop browsing link and it looked simple yet inviting – It offers a pleasant browsing experience that feels comfortable and easy to engage with for a while.
Online shoppers enjoy platforms that combine smart interface design with practical product selections, ensuring a seamless and efficient browsing experience every time Savvy Smart Shopping Experience Station Hub – providing clean layout structure, fast navigation performance, and organized categories that improve usability and user satisfaction significantly
Many digital shoppers enjoy platforms like premium trend centers that serve as a great place for discovering updated trends and modern product ideas daily Premium Trend Style Center Hub – The website is known for its modern design and structured browsing system that improves usability
Digital shoppers often highlight that engaging product selections make browsing more enjoyable and increase the likelihood of returning to a site, as seen on petal copper goods hub – the experience felt worthwhile since items looked appealing and made the platform feel like a place worth revisiting.
как отмыть траву с подошвы и как отмыть траву с белой подошвы эффективные способы очистки обуви
Душевой уголок AM.PM Like привлекает тем, как в нём сочетаются размеры, внешний вид и практическая логика. Даже выразительная по виду модель может оказаться спорной, если не совпадают размеры, способ монтажа и сценарий использования. Если эти моменты учтены заранее, выбрать получится не просто красивую, а действительно удобную модель на каждый день https://my-bathroom.ru/dushevye-ugolki/dushevoj-ugolok-am-pm-like-obzor/
Shoppers who value convenience tend to choose platforms that offer smooth navigation and easy checkout processes with minimal steps involved overall Smooth Purchase Point – The experience is designed to simplify buying decisions while ensuring users enjoy a fluid transition from browsing products to finalizing their orders efficiently
People exploring digital marketplaces prefer platforms that use modern layouts to help them find products quickly and complete purchases efficiently Fast Modern Buy Hub enhancing usability – It is often described as a user friendly site with clear navigation and organized product structure
People often gravitate toward e-commerce platforms that create a sense of openness and calm, especially when browsing wellness or handmade product collections with a clean visual presentation meadow inspired market view – The idea represents a soft, airy digital marketplace where natural goods and handcrafted items are displayed in a way that feels refreshing and easy to navigate.
While browsing through various websites, I stumbled upon browse blossom horizon and it felt neat and structured – The layout is clean and the design is simple, helping everything feel easy to explore without any clutter.
While exploring different online resources, I landed on take a look and thought it was a solid site, with information presented clearly and a layout that made browsing smooth and efficient.
Online visitors often value websites that prioritize usability and clarity because it makes browsing products and categories more efficient and enjoyable overall flash corner urban quick look the design feels structured and straightforward which helps users understand offerings quickly while maintaining a smooth and pleasant experience
Online buyers frequently prefer websites that highlight selected products rather than overwhelming them with too many options, improving overall shopping clarity and satisfaction 精选 Style Picks Zone – It offers a curated environment where stylish items are displayed thoughtfully, allowing users to browse quickly and enjoy a clean and efficient interface
While exploring different product listings online, I noticed discover this store – The browsing experience feels very pleasant, with clear navigation and fast page loading that makes it easy to move around the site.
During a light browsing session, I visited click to view and appreciated how well everything was arranged, creating a seamless and easy-to-follow experience.
Online visitors often mention that interesting product displays can make a site more memorable and encourage repeat browsing sessions, as seen on petalmarket copper collection – the browsing experience was enjoyable since items looked appealing and created a sense that the platform would be worth checking again later.
During casual exploration of shopping sites, I found a convenient platform especially when visiting Cove smart shopping list which offers organized categories and efficient browsing tools – The experience feels streamlined and user oriented allowing visitors to find products without unnecessary effort or confusion
Users frequently prefer shopping arenas that make cart handling simple and efficient, allowing quick purchase completion with minimal effort required Smart Cart Flow Simplified Arena Portal – offering fast navigation tools, clean interface structure, and optimized checkout experience that enhances usability significantly
Online shoppers who prefer streamlined e-commerce experiences often choose trend focused platforms like premium trend marts that provide useful items with simple user experience designed for quick browsing and efficient decision making Premium Trend Smart Mart Hub – The platform is appreciated for its clean layout, intuitive navigation, and fast loading pages that make product discovery smooth and stress free for everyday users
Many digital shoppers choose platforms that provide efficient cart systems allowing seamless shopping experiences from selection to checkout completion Modern Cart Control Hub making transactions easier – It is often recognized for its structured design and user friendly navigation system
During my exploration of digital marketplaces for budget-friendly shopping, I came across value deals explorer which simplifies browsing through categorized offers – Value market hub focuses on affordable items and easy navigation, ensuring users can find relevant products quickly while enjoying a smooth and efficient interface designed for easy product discovery.
People exploring digital marketplaces often appreciate floral-inspired environments that create a sense of calm while still maintaining vibrant and engaging product presentation across categories echopetal gift showcase hub – This reflects a delicate bazaar concept where decorative goods and artisan collections are displayed in a visually pleasing and naturally inspired layout.
While looking around different online platforms, I found shop link here and it felt quite refreshing in its presentation and style – The collection appears curated in a thoughtful way, making the browsing experience feel both distinctive and pleasant.
While reviewing digital storefront ecosystems and niche vendor platforms I came across FloraBrook shop catalog entry which provided structured categories and easy navigation across marketplace sections – I gathered useful insights while casually exploring its organized layout
During comparative analysis of online store checkout systems and their performance consistency, I noticed that reliability in cart operations directly affects user confidence and engagement, especially during high intent shopping sessions, which stood out when testing stable shopping cart – The experience is smooth and dependable, allowing users to navigate their cart easily while maintaining consistent performance across pages.
While checking out online shops for fun, I discovered visit this page and it felt very clear – The collection feels fresh and thoughtfully curated for shoppers, making browsing smooth, easy, and comfortable from start to finish.
Modern consumers tend to value platforms that keep them updated with current trends while offering a seamless browsing experience across devices Current Style Hub – The hub focuses on showcasing the latest product ideas with a responsive and organized interface designed for convenience
Many online visitors enjoy browsing platforms that showcase fashion trends in an organized and visually appealing way you can explore trending lifestyle shop offering a neat presentation of items that makes discovery easy and engaging for users exploring different styles – The browsing experience is fluid with clear visuals and simple navigation structure.
While searching for online shopping deals, I encountered explore this offer hub – The variety immediately stood out, making it feel like a worthwhile place to browse for good bargains.
Online users seeking modern retail solutions can visit Modern Basket Market which organizes products into a clean and user friendly structure – this helps simplify browsing and allows shoppers to enjoy a more efficient experience while exploring a wide variety of goods available across different categories.
Online shoppers often value platforms where products are presented in a way that encourages curiosity and repeated exploration over time, as seen on petal copper retail hub – browsing felt enjoyable because the items looked interesting and made the site feel worth revisiting for further exploration.
NeoSpin https://adventureallstars.tv/
Many users appreciate shopping platforms that focus on smart decision making, providing a wide range of products arranged in a simple and intuitive layout for easier browsing experience Smart Everyday Choice Market Bazaar – designed for smooth navigation, fast browsing flow, and organized product categories that enhance usability and overall shopping convenience significantly
During my evaluation of shopping platforms focused on usability, I came across a digital browsing solution online product finder that improves category organization and enhances user experience through structured navigation – Quick online market provides quicker access to product listings and ensures smoother browsing for users who prefer efficient and well-organized shopping environments.
Online consumers frequently prefer platforms that display exciting trends in a clean interface so they can explore products without unnecessary distractions Modern Trend View Hub improving experience – The website is recognized for its organized structure and visually appealing product presentation
While reviewing different resources online, I found a section that might be useful for broader browsing and research purposes, especially for general interest users who appreciate curated content, located here Quartz Orchard Collective homepage which can be explored further for additional context and navigation options within the site structure, offering multiple pages and categories that help users understand the layout and available materials – The page appears structured in a straightforward manner and seems easy to navigate for first-time visitors seeking organized information.
Digital shopping experiences are enhanced when websites provide clear navigation paths fast page loads and detailed product filtering options for users golden crest product hub access allowing them to quickly locate relevant items across extensive catalogs without wasting time or encountering unnecessary distractions during browsing – A product hub access point is particularly useful for large inventories where organization directly impacts user satisfaction and sales efficiency
Online browsing becomes more enjoyable when platforms incorporate natural aesthetics and sustainable themes that highlight handcrafted and eco-friendly product categories pine echo nature collective – The concept represents a forest-rooted digital space where products are presented with pinewood-inspired simplicity and environmentally conscious design principles.
Online buyers frequently prefer websites that combine visual appeal with simplicity so they can explore trending products without unnecessary distractions or delays Clean Style Trend Corner – The layout emphasizes clarity and ease of use, helping users navigate stylish items quickly and efficiently
I randomly clicked through a few pages and landed on explore this store which seemed neatly arranged – I found some appealing items here and the browsing experience was quite pleasant, calm, and easy to go through.
Online consumers frequently rely on premium trend zones that provide stylish trending products with clear and easy layouts for improved satisfaction Premium Trend Market Flow Zone Hub – The website is organized for fast navigation and easy product discovery
I was browsing through various e-commerce platforms when I came across modern deal marketplace which offered an organized catalog and smooth navigation making it easy to explore products – overall it felt like a decent shop with a few unique items worth noting.
I had been exploring multiple shopping websites when I came across see what’s here – It appears to be a decent shop with trendy items, and the pricing looks fair enough to attract casual online shoppers.
As I explored different online sites, I reached open this resource and found it quite useful, so I saved it for future reference and planned to revisit it when needed again.
Visitors to online marketplaces often note that interesting product variety plays a key role in keeping browsing sessions engaging and enjoyable, as seen on petal copper shop link – items were appealing and made the experience feel worthwhile, encouraging another visit to explore the catalog more thoroughly in the future.
During a relaxed browsing session, I found open shop page and it looked clean and simple to navigate – I came across a few interesting things that made the experience enjoyable and something I would suggest checking out.
Many bargain hunters prefer platforms that highlight discounts effectively, which is reflected in Smart Deal House Value Center – offering structured categories, simple navigation flow, and easy access to daily deals for convenient online savings experience today across all users.
While analyzing ecommerce platforms built for structured browsing and flexible product access, I observed that choice hubs help users navigate large inventories more effectively, especially when exploring diverse categories, which became evident when testing smart shopping variety hub – The interface provides rich product selection and allows easy switching between categories, creating a smooth and intuitive browsing experience overall.
People browsing for online shopping solutions appreciate platforms that reduce complexity and improve overall clarity in product presentation NextGen Market Place – The website is designed to support effortless navigation, ensuring users can quickly find items and enjoy a smooth shopping experience
During a routine browsing session, I checked out check it out and noticed it was a pretty decent platform overall, with sections that made exploring content feel enjoyable and easy to manage.
Online shoppers often value platforms that bring together trending products and ideas so they can easily stay updated with modern shopping choices Trend Center Navigator Hub making exploration easier – It is commonly described as a user friendly website with clear navigation and well structured product listings
Consumers who follow fashion and lifestyle updates often seek platforms that provide a mix of curated trends and new arrivals in an accessible format Trend Picks Hub – This version emphasizes curated selections of stylish goods while ensuring users can navigate easily and stay informed about the latest updates
Queen Win https://queenwincasinos.com/
Many users enjoy e-commerce platforms that feel structured and minimal, particularly when exploring lifestyle products inspired by frost and coastal design themes frost bay minimal goods portal – The concept reflects a cool-toned collective brand where curated items are presented in a clean and peaceful digital environment that enhances browsing ease.
As I moved through a few online pages earlier, I discovered visit spruce shop and it felt structured and clear – Nice variety of goods and everything looks well presented and organized, making the site easy to explore naturally.
I was exploring different online stores when I found have a look here – The user interface feels smooth and well-designed, making product discovery simple without confusion or difficulty navigating pages.
Many users browsing e-commerce platforms appreciate when the product selection feels fresh and inviting, making them want to explore further over time, especially on copper petal market view – browsing felt engaging because items seemed appealing and worth revisiting for a closer look at different categories and possible finds.
Many users appreciate prime value corners that deliver good value deals with practical and affordable choices for smooth transactions and browsing efficiency Prime Value Prime Corner Flow – The interface supports intuitive navigation and organized product listings
Shoppers who value efficiency often gravitate toward websites that minimize friction and provide clear pathways to explore different product options deal finder hub – The layout emphasizes usability and speed, ensuring that users can browse categories smoothly without unnecessary interruptions or delays in experience.
Online platforms that provide structured product categories and fast navigation are preferred by most digital shoppers, Golden Drift e-commerce entry as they reduce effort required to locate items and streamline the entire purchasing process effectively. This approach is widely considered beneficial for improving user satisfaction and conversion rates
E-commerce continues to advance with platforms like Ultimate Flex Shopping Zone offering highly adaptable product discovery tools – These systems prioritize user satisfaction through improved navigation, personalized recommendations, and flexible browsing structures that make it easier for customers to find exactly what they need in a simplified digital space.
базу данных купить [url=https://gobasego-3.ru/]базу данных купить[/url] .
Many shoppers prefer efficient discovery systems that reduce search effort, and Smart Pick Corner Browse Center – provides organized categories, fast loading interface, and curated product lists that make everyday shopping easier and more enjoyable for users across different needs.
During research into small business platforms and digital marketplace aggregators I encountered Stone collective showcase which organizes seller listings into sections, and my impression after browsing was that it is clean and easy to understand – overall it felt minimal and efficient
почему упала производительность ноутбука в играх и что делать для ее восстановления
People exploring online marketplaces frequently value platforms that provide fresh trending products updated regularly for easier browsing and selection Modern Trend Update Hub making shopping easier – The site is often recognized for its clean design and well structured categories that improve usability
Digital buyers often choose platforms that present fashion deals in a clean layout with easy filtering and search functionality Fashion Select Portal it helps users quickly identify relevant clothing options while maintaining consistent performance and a smooth browsing experience
Users frequently value e-commerce sites that present products in a way that feels engaging and encourages them to come back later, particularly on copperpetal goods center – browsing felt enjoyable since items looked interesting and gave a strong impression of being worth checking again.
Online shoppers who prefer speed and efficiency often look for platforms that allow them to browse items quickly without dealing with complicated layouts or slow loading pages Swift Goods Corner Hub – This platform focuses on delivering rapid product access through a clean interface that makes browsing smooth and convenient for users seeking fast shopping solutions
While checking out various online shops today, I came upon take a look here and it gave a fresh and polished impression – The items seem trendy and well curated, while the browsing experience felt fast and easy throughout.
While checking different shopping websites, I found visit this store page – The platform feels quite interesting, and the product images are clear and appealing, making browsing feel more engaging and visually informative.
While going through a variety of online stores, I encountered check crest items and it felt simple and clear – The overall feel was pleasant and browsing was quick and easy today, making everything easy to follow.
Online browsing becomes more engaging when platforms adopt strong winter mountain aesthetics, offering users a sense of calm structure while exploring rustic crafts and seasonal collections frostcrest peak lifestyle hub – It represents a mountain-themed marketplace identity where cool-toned decorative goods are arranged in a visually strong and immersive browsing structure.
As I navigated across various online pages, I encountered see this option and found it to be a clean and reliable-looking site that gave a solid impression during my casual browsing experience today.
Users often prefer shopping websites that provide a balanced combination of simplicity speed and well structured browsing experience Urban Pick Zone store link – this kind of design approach allows them to focus more on products rather than dealing with unnecessary visual complexity or distractions online
Many shoppers appreciate quality buy worlds offering a wide range of quality products making shopping simple and reliable for faster navigation Quality Buy World Clean Flow Hub – The interface provides minimal design and efficient browsing tools
Many online shoppers enjoy platforms that present curated trends in a simple format, and Smart Trend Arena Easy Flow Hub – provides organized listings, smooth navigation structure, and clear product categories that enhance usability and make shopping more enjoyable overall.
While exploring various online retail platforms focused on promotions, I came across a section labeled smart savings product hub – The interface presents tech deals in a clean layout that makes browsing easy, giving users a smooth experience while discovering well priced digital and online products across categories.
Online shopping experiences are improved when websites provide consistent layouts and easy access to different categories for better product discovery and comparison, DuneMarket entry point – The platform maintains a simple structure that helps users move naturally through pages while keeping the overall experience smooth and efficient
As I navigated through different online sources, I encountered go to this site and found it to be a nice site overall, with useful content that seemed worth revisiting later for deeper exploration.
Online users often value platforms that combine neon aesthetics with well structured shopping categories for a more engaging browsing experience Neon Flash Deal Hub enhancing usability – It is commonly noted for its bright interface and simple navigation flow
Online consumers today often search for platforms similar to quick cart selection hub – The cart choice store approach emphasizes fast navigation and intuitive browsing so users can quickly move from discovery to checkout without being overwhelmed by too many unnecessary steps or distractions
Digital shoppers often highlight that engaging product selections make browsing more enjoyable and increase the likelihood of returning to a site, as seen on petal copper goods hub – the experience felt worthwhile since items looked appealing and made the platform feel like a place worth revisiting.
During my browsing session for new stores, I discovered explore this page now – The platform gives a good initial impression, with organized categories and a browsing experience that feels intuitive and well-structured.
Shoppers who prioritize efficiency in online shopping tend to choose platforms that organize items clearly and allow quick transitions between sections Organized Quick Zone – The layout combines structure and speed to enhance the overall browsing experience
While checking out different craft websites, I came upon take a look and it gave a warm impression – The crafts look unique and interesting, making it definitely worth spending time exploring the collection thoroughly and calmly.
Digital users often prefer platforms that emphasize creativity and thematic depth, making browsing more engaging when exploring curated lifestyle and modern decorative products frostdune goods center – The concept represents a visually distinctive marketplace that blends frost and desert imagery into a unified and adventurous shopping environment.
casino https://seoanalizer.kseo.com.br/domain/nolimitcity.uk.com
Shoppers looking for updated wardrobe choices online usually value platforms that present items clearly and avoid cluttered design layouts apparel browsing site – the browsing flow feels efficient and well designed, making it easier for users to explore products and identify suitable fashion pieces
Digital shoppers often prioritize platforms that organize products clearly and reduce effort during selection, and one such example is Everyday Shop Point which highlights convenience while the service description explains how it helps customers quickly locate essentials and complete purchases in a smooth and structured online environment designed for regular household shopping
Shoppers who prefer simplicity often choose platforms that clearly present modern items, and Smart Trend Store Global Hub – provides structured browsing experience, intuitive navigation tools, and curated product updates that help users explore trends quickly and conveniently across different shopping needs today.
While exploring various online shops, I found browse unique crafts and it gave a pleasant and natural impression – The crafts appear authentic and nicely displayed, making browsing feel smooth and enjoyable.
Online consumers frequently rely on quality cart arenas that offer smooth cart experience with well organized categories and checkout flow for better satisfaction Quality Cart Arena Market Flow Hub – The website is structured for easy product discovery
Many shoppers enjoy websites that offer a neon inspired fun corner making it easy to find deals and products in a lively interface Neon Glow Fun Explorer Hub improving usability – It is frequently praised for its colorful layout and smooth browsing experience
During a light browsing session online, I visited click this collective and found the navigation smooth and the content clear, which made the browsing experience pleasant and straightforward overall.
Many people exploring digital stores tend to value platforms where products feel thoughtfully arranged and visually appealing for casual browsing, especially on copper petal goods site – the browsing experience felt enjoyable because the selection appeared engaging and gave a reason to return and explore more items later on.
Not sure if anyone else here is into discovering lesser-known shops, but I recently came across one that felt worth sharing in this discussion explore this hidden gem – There’s a noticeable sense of care in how everything is presented, and the products feel distinctive rather than generic.
анализ карточек маркетплейс [url=https://reklamnyj-kreativ18.ru]анализ карточек маркетплейс[/url]
super cherry [url=https://sheikhjameel.com/super-cherry-50-100-und-200-die-mittleren-varianten-im-detail/]super cherry[/url]
Online retail users expect seamless transitions between browsing pages and checkout steps, especially when exploring discounted or trending items quickly Rapid Purchase Stream – this platform ensures smooth performance and helps users move through the buying process without interruptions or unnecessary slowdowns affecting their experience
While browsing for interesting products online, I noticed discover this store – Everything appears nicely arranged, making online shopping easier and more enjoyable with a clean structure that helps users navigate effortlessly.
I came across something earlier that felt organized and worth sharing because of its interesting product presentation see this shop – The shop is nice overall, and the products seem interesting and worth checking out when you get a chance.
crazy time [url=https://wordpressbe.tomi.com/crazy-time-strategy-three-betting-approaches-that-control-your-variance/]crazy time[/url]
Users interested in fast shopping experiences typically look for platforms that combine curated content with responsive layouts for smoother browsing sessions Rapid Picks Zone – The platform focuses on delivering selected products in a fast loading environment that enhances usability
позиция карточки в выдаче [url=https://reklamnyj-kreativ19.ru]позиция карточки в выдаче[/url]
People browsing for artisan lifestyle goods often come across the Golden Fern Product Space section while navigating independent shop networks – it provides a clean and inspiring shopping experience centered on creative expression and carefully selected handmade pieces from various makers
While scrolling through various online shops I discovered a store that felt clean and aesthetic with well balanced visuals and gentle design tones silver petal goods hub – Beautiful collection here worth exploring for daily home inspiration today, making it easy to find creative home inspiration.
While passing time online and opening random pages, I reached visit this webpage and found the content pretty interesting, making it easy to go through and understand thanks to its simple presentation.
casino https://sisidigitaltools.com/website-reviewer/es/domain/playtech-demo.nz
During a casual online search across different marketplaces, I discovered explore blue petal and it stood out with a neat layout – The store design feels modern and comfortable to navigate, offering a smooth and enjoyable browsing experience overall.
пластиковые окна рулонные шторы с электроприводом [url=https://avtomaticheskie-rulonnye-shtory5.ru]пластиковые окна рулонные шторы с электроприводом[/url]
During a casual browsing session I found a website that felt serene and well structured with a clean interface and neatly arranged product sections that supported effortless navigation twilight oak shop – Store has calm aesthetic with easy browsing and clean layout, offering a smooth and relaxing user experience.
Digital users often enjoy e-commerce platforms that feel like exploring a magical frozen enclave, especially when browsing curated premium lifestyle and cozy themed product collections frost eden goods portal – The idea reflects a secluded winter sanctuary where products are displayed in a calm, elegant environment that enhances discovery and browsing comfort.
Online users often choose fashion platforms that simplify browsing and improve clarity, and Stylish Corner Style Flow Portal – features curated product listings, intuitive interface design, and fast navigation tools that help users explore stylish items with greater ease and efficiency across devices.
Online shoppers who value efficiency often look for websites that minimize clutter and present products in an organized manner arena fashion catalog – this structure helps improve browsing speed and ensures users can easily locate items without being distracted by unnecessary visual elements
Online visitors often mention that interesting product displays can make a site more memorable and encourage repeat browsing sessions, as seen on petalmarket copper collection – the browsing experience was enjoyable since items looked appealing and created a sense that the platform would be worth checking again later.
Many users appreciate quality cart stations that provide efficient shopping station systems with easy cart management and quick purchases for smooth transactions Quality Cart Station Prime Hub Flow – The website focuses on usability and clean design
During a casual browsing session, I discovered check out this page – Products appear stylish here, making it a nice and comfortable place to discover new things online without unnecessary hassle.
The online gambling landscape has evolved significantly over the past decade, moving from classic casino formats like slots and roulette to fast-paced interactive experiences.
One of the most notable innovations in this space is the “crash game” genre—minimalist yet highly engaging formats based on quick decisions and volatility here [url=https://www.barbadossothebysrealty.com/story/astronaut-game]Astronaut Crash Game[/url]
Among these, the Astronaut game stands out as a modern crash-style game that combines clean design and unpredictable multiplier systems.
This article provides a full informational breakdown of the Astronaut crash game, its mechanics, appeal, risks, and its place within the broader online casino ecosystem.
It is intended for readers aged adult users interested in gaming systems who are interested in analyzing modern gambling formats from an informational perspective.
How the Astronaut Crash Game Works
Astronaut is a real-time betting game where a multiplier rises gradually during gameplay while an animated astronaut travels upward in space.
The core idea is simple: the longer the astronaut travels, the higher the multiplier becomes.
Players typically enter a wager prior to launch and must decide when to secure their multiplier before the system randomly stops the ascent.
If the player cashes out in time, they receive a payout based on the multiplier at that moment.
If the game crashes before cash-out, the stake is lost.
Main Game Loop
At its heart, Astronaut follows a straightforward cycle:
A round begins and players place a stake.
The astronaut launches and a multiplier starts increasing (e.g., low to high multipliers).
At any moment, the player may choose to cash out.
If the player cashes out before the crash, the payout equals stake ? multiplier.
y=mx
m
If the crash occurs before cash-out, the stake is lost.
The unpredictability of when the crash happens is what defines the game.
Unlike traditional games based on fixed probability systems like roulette or blackjack, crash games rely on probabilistic systems.
The Appeal of Crash Games Like Astronaut
1. Simplicity
Astronaut does not require complex rules or experience.
The interface is typically minimal: a rising multiplier, animation, and a cash-out button.
2. Fast Rounds
Each round lasts brief interactive cycles, creating constant action.
3. Decision Pressure
Players face a choice between early secure wins and higher risk rewards.
4. Visual Theme
The Astronaut theme adds visual engagement to a mathematical system.
Game Risk Dynamics
Crash games are considered unstable outcome-based games.
Outcomes may include:
unexpected end points in rounds
Long streaks of minimal outcomes
Occasional rare big wins
The psychological tension often comes from the idea of delaying cash-out decisions.
Behavioral Triggers
Anticipation Effect
As the multiplier increases, excitement rises.
Near-Miss Effect
Players often experience timing mistakes close to crashes, which increases engagement.
Perceived decision power
Although outcomes are random, players feel they can influence results.
Rapid Feedback Loop
Each round provides instant results, reinforcing continued play.
Comparison with Other Crash Games
Astronaut belongs to a category including other multiplier crash systems.
Compared to earlier versions, Astronaut features:
improved visuals
Space-themed narrative
clean interface
Common False Beliefs
Some common beliefs include:
“A high multiplier is due after low rounds”
“winning streaks can be predicted”
However, these assumptions do not reflect how independent probability functions operate.
Each round is separate.
Responsible Perspective on Gameplay
Astronaut operates within a gambling framework.
Key points:
losses are possible at any time
No guaranteed earnings exist
results vary widely
It should be treated primarily as a game of chance, not a method of income.
Why Astronaut Fits Modern Casino Trends
Modern trends include:
Mobile-first design
Gamification
Short-form engagement
community-driven gameplay
Summary
Astronaut is a multiplier-based gaming experience combining interactive gameplay and randomness.
Its appeal lies in risk-reward balance.
However, it remains a non-skill-based environment, where results cannot be reliably predicted or controlled.
As crash games continue to evolve, Astronaut represents a clear example of how online casino formats are becoming more modern and gamified, while still relying on core randomness principles.
Came across something interesting while scrolling earlier and thought it might be relevant to share here with everyone reading this thread discover this shop – The overall feel is quite unique, and it seems like effort was put into selecting items that stand out individually.
During research into modern online retail systems and plugin environments, I discovered cart zone explorer which focuses on simplifying store creation workflows – it provides helpful insights into managing products, improving storefront structure, and supporting users who need flexible tools for building efficient and easy-to-navigate e-commerce platforms with consistent performance.
While looking through various online stores I came across something that immediately gave a peaceful and well organized impression radiant cove selection and it felt nicely curated with a soft visual tone that made browsing feel effortless, – Everything appears carefully arranged, offering a calm shopping environment where each section flows naturally into the next without confusion or visual clutter.
Online buyers typically prefer websites that reduce the number of steps needed to select items and complete transactions smoothly and securely Fast Choice Market – The system focuses on simplifying both browsing and checkout, ensuring users can shop without unnecessary delays
As I explored multiple online platforms, I came across view items now and it gave off a clear and tidy vibe – The clean layout and simple navigation made browsing feel smooth, easy, and enjoyable throughout.
casino https://report.nadvertex.com/en/domain/playtech-demo.nz
While going through different handmade shops, I encountered check forge items and it felt calm and neat – The items appear creative and the browsing experience was smooth overall, making it easy to browse without feeling overwhelmed.
While casually searching the internet I discovered a site that felt elegant and simple with a clean interface and organized categories silver petal goods store – The emporium feels well organized with many appealing items available now, making browsing feel clear and efficient.
Online shoppers often enjoy platforms where browsing feels dynamic and the product selection encourages curiosity and repeated exploration, as seen on petalmarket copper store – items were appealing and made the browsing experience feel enjoyable enough to return for another look in the future.
People exploring modern e commerce platforms frequently search for stores that combine variety and simplicity while offering creative products for everyday use and gifting purposes Fern Marketplace Portal – It offers a well structured shopping experience with balanced selection and an easy to navigate layout that enhances overall user satisfaction during browsing
Online retail environments are becoming more focused on affordability and simplicity, allowing users to quickly locate relevant deals across different product categories Budget Finds Portal this reflects how digital shopping platforms evolve to support cost conscious users who want reliable access to everyday savings and special promotions
Many digital shoppers appreciate platforms that combine winter and floral themes, offering a visually unique browsing experience when exploring curated artisanal goods and seasonal decorative collections across organized product categories frostgarden floral winter portal – It suggests a hybrid aesthetic where icy frost meets blooming garden elements, forming a gentle and artistic marketplace atmosphere designed for handmade products and seasonal décor lovers.
Users browsing e commerce fashion sites often prefer layouts that are minimal, organized, and easy to understand while shopping online today experience UTC online showcase – the platform offers a smooth browsing experience, ensuring that users can explore collections efficiently without unnecessary distractions or clutter during usage sessions overall easily
I was casually exploring online shops when I noticed take a look here – The selection seems good, and it looks like they keep updating their inventory with new and fresh items that are easy to discover while browsing.
While exploring different online stores I came across a shop that felt modern yet calm with a balanced layout and intuitive category browsing system for simple discovery twilight oak collection – Store has calm aesthetic with easy browsing and clean layout, making product exploration feel natural and stress free.
Many shoppers appreciate quality trend centers providing trend focused browsing with updated products and clean browsing design for faster navigation Quality Trend Center Clean Flow Hub – The interface provides minimal design and efficient browsing tools
I came across a store while browsing that felt very clean and simple which made it easy to understand the layout quickly radiant essentials guide – Browsing this store was easy and the items seem quite useful especially for practical everyday usage and simple lifestyle needs.
I was just casually exploring the internet when I came across something that felt simple yet well put together, so I thought I’d mention it check this place out – Everything looks clean and structured, which makes browsing feel effortless and smooth.
casino https://theavtar.in/read-blog/147915_traditional-board-activities-on-32-red-casino.html
As part of my browsing routine, I came upon visit this link and noticed how the layout supports readability, with a clean structure that allows information to be understood quickly and without effort.
Users exploring online marketplaces frequently enjoy when product listings feel fresh and visually interesting, encouraging longer browsing sessions and return visits, particularly on copperpetal market access – browsing felt satisfying since items appeared intriguing and gave a reason to check the site again in the future.
During my usual browsing routine, I encountered check trader page and it immediately felt clean – The products are nicely selected and browsing here was simple and enjoyable, making navigation easy and pleasant.
Online users often appreciate e-commerce platforms that feel inspired by forest groves and rustic craftsmanship, making browsing more immersive when exploring curated handmade décor and artisan lifestyle products frost grove wooden craft hub – The idea represents a cold woodland marketplace where handcrafted goods are displayed in a natural, earthy environment that highlights artisan skill and rustic design values.
During my browsing session for new stores, I discovered explore this page now – The website appears modern, and I had a pleasant time scrolling through different product sections today because everything felt smooth and easy to follow.
I was casually browsing online when I found a store that felt minimal and user friendly with a clean interface and well arranged categories silver petal essentials – The store layout is simple and makes browsing very smooth experience, helping create a relaxed browsing environment.
I randomly clicked through a few pages and landed on see this page which seemed nicely arranged – The items look thoughtfully selected, creating a calm and smooth browsing experience overall.
Shoppers frequently note that browsing different collections on the site feels fast and stable even during peak usage hours when traffic is higher than usual trend hub express making the overall experience feel reliable and convenient as users can explore multiple categories without facing lag or delays in page transitions or product loading.
I was exploring different websites when I stumbled upon something that felt visually clean and easy to understand right away radiant collective field store – The design is clean and navigation is simple, making the browsing experience pleasant and comfortable today overall.
Online shoppers who follow modern product updates often explore quality trend stations that act as reliable trend stations showcasing modern items and frequent product updates designed for smoother browsing and better decision making Quality Trend Station Flow Hub Explorer – The platform is appreciated for its clean layout, structured navigation, and fast loading interface that helps users quickly discover updated products without confusion or delays
I don’t usually share links, but I found something earlier that felt interesting enough to mention here for anyone curious check this place out – There are a few unique items, and I’d probably revisit it again soon.
In between checking multiple sites, I encountered visit this page – The design feels neat and browsing through it is straightforward and comfortable.
Many visitors to digital shops appreciate when the variety of items creates a sense of discovery that makes browsing more enjoyable, especially on copper petal showcase hub – the experience felt engaging because products seemed interesting and made the platform feel worth exploring again later.
I randomly clicked through a few pages and landed on explore this store which seemed well arranged – Everything appears well structured and easy to browse through quickly, giving a calm and easy browsing experience.
I had been browsing multiple websites when I noticed visit this page – My experience so far is nice, with clearly arranged items and descriptions that are informative and easy to understand.
Digital users often prefer platforms that evoke peaceful coastal winters, especially when browsing curated lifestyle goods and aesthetic seasonal product collections frostharbor artisan goods hub – The concept represents a harbor-inspired marketplace where curated items are displayed in a cool-toned, structured environment influenced by frozen coastal scenery.
Fashion enthusiasts who spend time exploring online shops typically appreciate sites that highlight trending apparel in a simple and accessible way fashion station browse page and the store gives a decent impression with plenty of fashionable items that seem relevant for current style trends and casual browsing interest
I was exploring various websites when I found one that felt stylish and modern with a clean layout and organized browsing structure silver sprout goods collection – Nice selection of products here with a fresh and modern look, helping users browse comfortably.
While casually exploring the internet I found a site that had a slightly unconventional feel compared to normal online shops radiant grove goods view – It has an interesting selection that feels different from usual online stores, making it stand out in a subtle but noticeable way.
Many online users appreciate stores where the product variety feels appealing enough to make them consider returning later for another look, especially on copper petal browse store – browsing was enjoyable because items seemed interesting and left a positive impression that encouraged future visits.
Online users often select quality trend zones because they provide a nice zone for product discovery with simple navigation systems that make browsing easy and efficient Quality Trend Zone Navigator Flow Hub – The website is optimized for clarity and structured transitions
In between checking various online stores, I encountered browse this page now – The site has a calm and cozy feel, with a layout that is clear and easy to follow.
I was casually looking through different online options when I found something that felt relaxing and pleasant to browse see this tranquil site – The items appear to be of good quality, and the presentation has a soothing, calm style.
As I explored a variety of websites during some free time, I came across go to this link and it looked fairly decent on the surface, making me consider returning in the future to see more of what it offers.
During my usual browsing routine, I encountered shop this page and it immediately felt well arranged and clear – I liked browsing here since everything is organized and visually appealing, making it pleasant to scroll through without effort.
In between checking multiple online platforms, I encountered browse trendy value site – I took a quick look, and the design feels clean and simple, making browsing easy enough overall.
During my online browsing time, I stumbled upon browse essentials items and it felt easy to use – The essentials appear practical and nicely displayed across the site, making everything feel smooth and user-friendly.
During my browsing session for new stores, I discovered explore this page now – The shop feels legit, with a clean layout and an impressive product variety that makes the browsing experience smooth and enjoyable.
Users exploring modern e commerce platforms often appreciate interfaces that feel clean, structured, and easy to navigate while browsing different product categories Vivid Cart Experience Hub – The platform delivers a smooth shopping flow with intuitive navigation and visually balanced layout design that makes browsing products simple, enjoyable, and efficient across all sections and categories without confusion or delays
Many shoppers enjoy online marketplaces that feel grounded in seasonal transitions, especially when browsing curated rustic décor and handcrafted lifestyle products frostharvest goods seasonal showcase – The concept represents a hybrid seasonal identity where goods are arranged in a visually soft, nature-inspired digital space blending harvest and winter aesthetics.
Many visitors exploring online marketplaces often mention that browsing feels pleasantly engaging when product selections are varied and visually appealing, especially on platforms like copper petal hub – the overall experience felt enjoyable since items looked intriguing and encouraged further exploration during the session with a sense of curiosity about what else might be available.
I stumbled upon this website while browsing randomly and it gave off a clean and minimal impression right away radiant maple shop hub – I found some unique items while exploring this site earlier today, and the experience felt smooth and easy to go through.
I had been looking through several platforms when I noticed open goods corner – Everything appears properly organized, and browsing feels smooth and convenient overall.
During a casual browsing session I came across a website that felt minimal and earthy with a balanced interface and stone themed design elements stone light shop – Stone inspired designs give this shop a unique aesthetic appeal, making the overall look feel creative and distinctive.
Users exploring e-commerce platforms frequently prefer rapid buy markets that act as fast shopping markets with easy access to everyday useful products designed for efficiency and clarity Rapid Buy Market Style Flow Hub – The interface is modern, intuitive, and optimized for seamless browsing
Online visitors who enjoy browsing clothing categories frequently look for platforms that make it easy to discover new and stylish outfits quickly station style marketplace and this store appears to provide a variety of trendy choices that look suitable for casual fashion exploration and general shopping interest
I ended up finding something while browsing that felt straightforward and worth sharing because of how easy it was to navigate explore this collection – The layout is clean, and the selection is presented in a simple and organized way.
In the middle of checking different shopping platforms, I came across open urban arena – The layout feels simple and clean, making browsing through pages smooth and natural without any issues.
I had been browsing multiple shopping sites when I came across see this collection – The layout is very structured, making items quick and easy to find while keeping the browsing experience smooth and efficient.
During a casual online search across different marketplaces, I discovered explore cloud meadow and it stood out with a soft clean design – The calm layout made browsing feel relaxed and quite smooth, creating an easy and enjoyable experience overall.
Users interacting with modern websites often appreciate platforms that combine minimal design with high performance for a smooth browsing experience CleanVivid Trend Center – It offers a fast and well organized interface where pages load quickly and content is arranged clearly, allowing users to browse efficiently while maintaining focus and comfort throughout their session
Many visitors exploring online marketplaces often mention that browsing feels pleasantly engaging when product selections are varied and visually appealing, especially on platforms like copper petal hub – the overall experience felt enjoyable since items looked intriguing and encouraged further exploration during the session with a sense of curiosity about what else might be available.
Many online shoppers prefer e-commerce platforms that feel warm and inviting, especially when browsing curated winter décor and artisan lifestyle collections frostlane cozy craft portal – The idea suggests a boutique emporium where products are presented in a calm, seasonal structure emphasizing handcrafted quality and aesthetic comfort.
I found a website while exploring online that felt visually clean and easy to understand at first glance radiant pine browse collection – Everything looks well organized and easy to browse without confusion, making it feel simple and efficient to use overall.
I spent some time checking out various online stores and eventually found browse this store which had a very neat presentation – The site design feels modern and smooth, creating a browsing experience that is comfortable and easy to enjoy at a relaxed pace.
During a casual browsing session where I was exploring different stores, I came across visit this collective shop – Everything runs smoothly, pages load fast, and the content feels updated and fresh throughout the experience.
During a casual online search session, I ended up exploring check this site and found the layout to be quite clean and accessible, making it easy to follow along and quickly grasp what was being presented.
While browsing online I came across a store that felt clean and practical with a simple layout and well structured categories that made navigation smooth and easy sun haven essentials hub – Essentials here feel practical, useful, and nicely presented for shoppers, giving a clear and simple browsing experience overall.
Online consumers frequently choose rapid cart centers that feature quick cart systems designed for smooth and efficient checkout processes improving clarity and speed during online purchasing Rapid Cart Center Express Hub Access – The interface ensures fast transitions between cart and payment stages with minimal complexity
Online deal seekers frequently look for platforms that specialize in lower priced goods while still maintaining a decent level of product variety and usability discount factory shop link and this store appears to align with that expectation by offering budget oriented choices that feel practical for everyday shopping needs and general household purchases.
While going through different websites today, I stumbled upon something that felt solid enough to share in this discussion visit this online store – The overall presentation seems dependable, and the structure looks well organized and carefully arranged.
While exploring multiple online options, I discovered see this urban buy site – The layout is organized well, and I could find things without much effort throughout browsing.
While browsing for interesting products online, I noticed discover this store – It’s a decent store, and browsing feels smooth while categories are easy to navigate and well structured for users.
Many online users appreciate stores where the product variety feels appealing enough to make them consider returning later for another look, especially on copper petal browse store – browsing was enjoyable because items seemed interesting and left a positive impression that encouraged future visits.
Modern web visitors frequently value platforms that offer straightforward navigation and consistent structure across all pages for improved usability and engagement TrendEase Vivid Station – It delivers a user friendly browsing environment with fast response times and simplified layout design that helps users access content easily while maintaining a clean and organized digital experience throughout the site
During my online browsing time, I stumbled upon browse petal items and it felt very delicate – The aesthetic feels soft and pleasant, and the items look carefully chosen, making browsing simple and stress free.
скачать с ютуба видео в хорошем качестве [url=https://skachat-video-s-youtube-12.ru]скачать с ютуба видео в хорошем качестве[/url]
Many online shoppers appreciate marketplaces that combine eco-conscious design with natural aesthetics, especially when browsing curated handmade crafts and lifestyle goods frostmeadow calm nature portal – It represents a peaceful winter meadow brand where goods are arranged in a structured, soothing environment inspired by frost and greenery.
While exploring online I found a shop that felt quick and reliable in performance, making it easy to browse without delay radiant shore view hub – Pretty smooth experience overall, and pages load quite fast here, keeping everything responsive and pleasant to use.
In the middle of comparing various online shops, I came across open this flower market – It has a pleasant atmosphere, and browsing different sections feels straightforward and well organized.
Modern shoppers appreciate streamlined websites with intuitive navigation and layout designs today Seamless Shop Arena – Organized content presentation enables faster browsing decisions by clearly separating product categories and allowing users to efficiently locate items without confusion or excessive scrolling effort
When reviewing multiple online shopping resources for better deal discovery, I encountered market deals explorer which organizes promotional listings in a user-friendly way – this shopnetmarket.shop system supports smooth navigation, allowing users to scan offers efficiently and compare products without unnecessary complexity or time-consuming searches across different sections of the platform.
bluefocus.click – Blue focus provides clear direction and strong digital strategy guidance
Many digital users enjoy rapid cart hubs featuring central hub design for fast cart handling and convenient online shopping enhancing everyday shopping convenience Rapid Cart Hub Flow Smart System – The website is appreciated for its clean structure and usability
While exploring multiple online options, I discovered see this cart arena hub – It offers a pretty decent experience, and the layout feels modern and easy to navigate.
Users exploring online marketplaces frequently enjoy when product listings feel fresh and visually interesting, encouraging longer browsing sessions and return visits, particularly on copperpetal market access – browsing felt satisfying since items appeared intriguing and gave a reason to check the site again in the future.
Many users browsing e-commerce sites prefer layouts that reduce clutter and make product discovery more enjoyable and efficient Goods value finder hub – navigation remains smooth and categories are easy to understand which helps improve overall usability for visitors online experience.
I stumbled upon something today that felt tidy and well structured, which made it stand out enough to share visit this shop – It’s a nice store, and I really like how everything is clearly sorted into categories.
While casually searching the internet I discovered a site that felt cheerful and simple with a light layout and organized categories sun meadow goods store – Store feels bright, welcoming, and easy to browse through categories, helping users find items quickly.
As I browsed through several websites earlier, I came across open collection and it looked simple and tidy – The selection here appears decent, with products that feel quality based and are nicely displayed for easy browsing online.
While comparing multiple e-commerce platforms I observed usability differences and found BestTrendStation browse page – Great browsing flow, content is clear and visually quite appealing, ensuring a smooth and structured navigation experience that makes it easy to move between categories efficiently
While browsing through several online stores earlier today, I came across cloud petal market hub and it immediately felt welcoming and simple – The marketplace looks friendly and easy to navigate through different sections, making the experience smooth and comfortable overall.
During random browsing I came across a site that felt visually unique with a subtle artistic glow in its presentation shadow glow collection hub – This shop has a cool aesthetic and includes interesting product choices that make it more engaging than typical online stores.
I was looking through various online stores when I noticed check this peak page – The site is very user friendly, and navigation feels easy, which made browsing around quite enjoyable.
Many digital shoppers enjoy platforms that combine floral elegance with frosty seasonal themes, especially when browsing curated decorative goods, aesthetic gifts, and lifestyle products presented in organized, visually calming layouts frostpetal floral goods portal – It suggests a delicate emporium identity where products are arranged in a soft, elegant structure inspired by winter frost and blooming floral design elements.
rent a yacht in Montenegro luxury Montenegro yacht charter
Users interacting with online stores often prefer fast and clear navigation systems that allow effortless movement between sections and product listings without disruption where Browse Cart Arena – this design supports efficient exploration by maintaining consistency across pages and helping users stay oriented during their shopping journey
For individuals who regularly shop online and want to maximize savings, platforms like the one referenced here can be quite helpful promo discovery guide – it is generally seen as a convenient way to browse various promotional listings, allowing users to quickly scan available deals without unnecessary distraction.
As part of my browsing routine, I encountered visit this link and during a quick visit, I could tell that the information was useful and the layout was nicely put together.
Many visitors exploring online marketplaces often mention that browsing feels pleasantly engaging when product selections are varied and visually appealing, especially on platforms like copper petal hub – the overall experience felt enjoyable since items looked intriguing and encouraged further exploration during the session with a sense of curiosity about what else might be available.
While exploring different websites for ideas, I came across visit this cart zone page – Everything loaded quickly, and I liked how it made browsing more enjoyable and easy to use.
[url=https://kabel-cena-za-metr-1.ru]кабель цена за метр[/url]
Many shoppers enjoy rapid cart solutions offering helpful solutions for cart management and streamlined buying experience overall, making online shopping more convenient, predictable, and user friendly Rapid Cart Solutions Easy Flow System – The platform is designed for simplicity, fast performance, and smooth checkout handling
While exploring various sites today, I discovered something that felt authentic and reliable enough to mention in this discussion browse this genuine site – It looks like a trustworthy place, and everything feels thoughtfully arranged with honest presentation.
Online shoppers frequently mention that well designed product galleries with high quality images help them feel more confident while browsing different categories and comparing available items across listings vivid product display center and the overall layout supports easy viewing, making it simple for users to focus on product details while enjoying a clean and visually attractive shopping environment.
светильники иркутск https://svetotekhnika-1.ru
While testing various e-commerce platforms I focused on interface flow and usability and found Best Choice Online catalog portal – Helpful site overall, makes finding items quick and convenient today, with an organized layout that allows users to explore products easily without unnecessary complexity
While exploring online I stumbled upon a marketplace that felt clean and structured with a modern interface and smooth browsing flow sun petal view hub – Market has good variety and items seem fairly well priced, ensuring a comfortable and easy experience.
While checking out different online stores, I came upon take a look and it gave a calm impression – The design is simple and neat, making browsing feel quick and comfortable, allowing everything to be explored at an easy pace.
нарколог на дом анонимно [url=https://narkolog-na-dom-voronezh-11.ru]нарколог на дом анонимно[/url]
While browsing for new online stores, I discovered browse flower store – The site looks clean and well organized, which makes finding what you need quick and easy.
частный нарколог на дом [url=https://narkolog-na-dom-voronezh-12.ru]частный нарколог на дом[/url]
I was casually browsing online when I discovered a shop that felt soft in design and easy to move through without distraction silk dune essentials – I enjoyed browsing here, as the items appear stylish and reasonably presented, making the experience feel comfortable and enjoyable.
нарколог на дом цена [url=https://narkolog-na-dom-voronezh-10.ru]нарколог на дом цена[/url]
Many digital consumers prefer soft and minimal retail platforms, especially when browsing curated floral-inspired décor, aesthetic gifts, and lifestyle product collections frostpetal winter retail hub – This reflects a gentle marketplace identity where products are presented in a calm, floral-winter inspired structure designed for relaxed browsing.
Shoppers increasingly prefer organized e-commerce layouts that reduce friction during browsing CartFlow Market Center provides structured category access while ensuring users can move between sections smoothly with improved usability and a more intuitive shopping experience designed for everyday customers overall experience.
During a casual online search across different websites, I discovered explore petal shop and it stood out with its soft visual style – The theme feels clean and welcoming, with a gentle design that makes browsing feel comfortable and visually soothing throughout.
During exploration of user-friendly e-commerce websites, I found easy shopping portal that simplifies product discovery – Simple shopping experience with clear layout and easy product access enhances usability by offering clear categories and straightforward navigation, allowing users to shop efficiently without unnecessary complexity or overwhelming design elements.
In the middle of comparing different platforms, I came across check this flash corner page – The site feels light and responsive, making it a nice place to explore without complications.
Shoppers today often rely on rapid goods centers that provide fast access to goods with well organized categories and listings making browsing more efficient and stress free Rapid Goods Center Select Flow System – The platform is structured for simplicity and easy navigation across categories
Creative tech teams often need platforms that support both visual excellence and technical efficiency for building modern web experiences SkyStudio Creative Flow – It provides a seamless combination of elegant design and fast performance, ensuring users can work within a visually refined environment that enhances productivity and delivers consistent professional quality across all interactions
I wasn’t searching for anything specific, but I ended up discovering something that felt pleasant to browse and worth sharing here explore this prism shop – The interface is clean, and navigation feels simple, which makes the overall experience enjoyable and stress-free to go through.
Many users exploring online stores value websites that allow quick and smooth navigation, ensuring that browsing remains efficient even when switching between different product sections vivid shopping flow hub and the interface feels polished and responsive, helping users scroll through content effortlessly while maintaining a pleasant and consistent browsing experience throughout.
I was browsing late in the evening when I found a site that felt calm and minimal with a clean design and structured product display sun petal collection hub – Simple store design makes shopping here quick and enjoyable experience, offering a smooth and relaxing shopping flow.
While exploring different online shopping platforms today just to compare usability and layouts, I came across something in the middle like check this pine collective – The browsing experience feels nice overall, with everything structured clearly and easy to follow without confusion or clutter.
While exploring different online stores, I found see this shop and it gave a polished impression – Products appear well organized and easy to find on this site, making browsing clean, simple, and enjoyable.
During my usual browsing I found a store that felt like a calm marketplace environment with a straightforward layout design silver bay collection hub – The marketplace feel is nice overall, and the variety of products available here is decent enough to keep browsing interesting.
Online users often appreciate platforms that evoke strong forest imagery, especially when browsing curated rustic décor and eco-conscious handmade products frost pine design hub – It suggests a pine-inspired marketplace where goods are displayed in a structured, earthy environment focused on sustainability and artisan values.
Shoppers enjoy online environments that prioritize clarity and smooth navigation across all product categories to improve browsing satisfaction overall ZoneNavigator Cart Pro – the platform delivers organized product access and efficient browsing flow, making shopping easier and more enjoyable for all types of users regularly.
Digital consumers increasingly value fast and intuitive shopping environments that reduce friction during the entire purchase journey Express Cart Link this platform supports efficient browsing and clear navigation – it helps users find products quickly while maintaining a smooth checkout experience that feels natural and effortless across all devices
вызов нарколога на дом анонимно [url=https://narkolog-na-dom-voronezh-13.ru]https://narkolog-na-dom-voronezh-13.ru[/url]
In the middle of checking different shopping platforms, I came across open flash hub – Everything feels clean and well structured, and the content is easy to follow along without unnecessary complexity.
Many digital shoppers appreciate rapid goods corner systems offering convenient corner for quick shopping and simple product discovery today, helping streamline the buying process and improve overall satisfaction ClearCart Goods Corner Navigation Portal – Focuses on structured flow and fast access to product listings
While scrolling through different sites earlier, I came across something that felt interesting and easy to browse, so I’m sharing it here check out this site – I enjoyed exploring it, and it seems like a reliable shop with a simple and structured layout that feels easy to use.
Digital shoppers looking for curated agricultural collections often seek platforms that balance product diversity with clean and accessible design Golden Crop Collection Shop – It offers a curated selection of harvest inspired products displayed through a user friendly interface designed to enhance browsing comfort and clarity
While browsing through several online shops earlier today, I came across amber petal finds and it immediately caught my attention with its variety – The products seem interesting and a bit different, and browsing here turned out to be surprisingly fun and enjoyable overall.
During my usual browsing routine, I stumbled upon see ridge collective – The interface looks modern, and everything responds well, making navigation simple and comfortable.
Many online shoppers prefer websites that present products in a well organized layout, making it easier to move between sections and discover items without feeling overwhelmed by cluttered design or confusing navigation paths cart zone shopping portal and the arrangement of categories appears neat and user friendly, helping visitors quickly understand where everything is located while browsing different sections of the store.
While reviewing the site and scanning its content, I noticed that learn more here integrates well into a clean layout – discovered it today, and everything looks informative, engaging, and neatly presented overall.
During my usual browsing session, I encountered check goods page and it immediately felt easy to use – The interface is nice and clean, making browsing smooth and straightforward overall, helping everything load and display clearly.
During random browsing I stumbled upon a site that felt minimal and clean with a layout that made everything easy to find silver crest selection – The products look reliable and descriptions are clear and helpful overall, giving a smooth and informative browsing experience.
I was exploring various websites when I found one that felt modern and structured with a clean layout and organized product sections sun spire goods collection – Collective offers interesting products and overall pleasant browsing experience today, helping create a smooth user journey.
Smart shopping arenas help users make better purchasing decisions by presenting comparisons discounts and organized product listings in one accessible location Smart Shopping Arena It creates an interactive environment where shoppers can evaluate offers efficiently compare options and choose the best value products without unnecessary browsing complexity or confusion
Many online users enjoy platforms that evoke ocean serenity in winter, especially when browsing curated seaside décor, minimalist lifestyle goods, and cool-toned home products frost shore craft portal – It represents a coastal winter identity where goods are arranged in a visually soft structure inspired by calm seas and frosty shoreline aesthetics.
While reviewing online marketplace designs for usability and layout quality, I found a platform that combined simple navigation with a broad selection of products across categories BuyZone product marketplace hub placed within the page flow – The experience feels organized and efficient, allowing users to browse easily while exploring a diverse range of items without difficulty.
As I examined the interface and content arrangement, I noticed that this page here contributes to a simple experience – I checked it out quickly, and it seems like a neat and useful site overall.
During my usual browsing routine, I stumbled upon see flash market – The structure is good overall, and nothing feels confusing since everything is easy to see and follow.
After analyzing how the content is arranged, I found that see this website fits into a well-organized design – clean website overall, and it feels modern and easy to browse around today.
Users browsing modern websites typically value clean design, responsive interfaces, and navigation that feels smooth and easy to use on every device UltraWave Flow Interface – UltraWave offers a clean and modern layout where browsing is smooth and easy, ensuring users enjoy a comfortable and intuitive experience.
I had been exploring different sites when I found open collective shore – The content is simple and clearly presented, making the first impression feel smooth and easy to process.
As I continued browsing through different areas of the site and examining its design, I realized that explore this page contributes to a seamless experience – everything feels smooth, and the structure is clear and easy to understand.
I like sharing interesting finds occasionally, and this one stood out because the collection felt worth revisiting later browse this oak market – It’s an interesting collection, and I might come back again to explore more items when I can browse more carefully.
People interacting with online platforms typically expect fast navigation, clear design, and well-organized content that helps them quickly find relevant information SummitMedia Access Portal – SummitMedia delivers a smooth and clean browsing experience where users can explore content easily while enjoying a visually structured and modern interface design.
частный нарколог на дом [url=https://narkolog-na-dom-voronezh-14.ru]частный нарколог на дом[/url]
Shoppers exploring e-commerce platforms often find interesting items that make them want to return later and take a closer look at additional listings and updated collections vivid trend catalog page and the browsing experience feels enjoyable enough to encourage repeat visits for further product discovery and exploration of new arrivals.
While checking out different online stores, I came upon take a look and it gave a calm impression – The variety is good and everything looks arranged in a clear way, making it easy to explore without confusion.
While casually browsing different websites I found something that felt modern and smooth with a clean and organized structure silver dune browse collection – The site feels modern and easy to navigate for new users, making it simple to move between pages comfortably.
Many online shoppers today look for platforms that combine affordability with modern design and easy navigation for daily purchases Stylish Deals Hub This platform aims to bring curated discounts and a smooth browsing experience so users can quickly find trending products and seasonal savings without unnecessary complexity or confusion
Users searching for reliable product depots online often prefer platforms that store varied items in organized ways and they visit the Horizon Goods Depot which provides a structured marketplace with a wide selection of goods and an intuitive browsing system that helps users quickly locate products across multiple categories.
Shoppers looking for convenience often rely on platforms that centralize basic household items so they can complete everyday purchases without unnecessary complications or delays home supplies portal – Such services are valued because they simplify access to common goods and make it easier for users to manage their daily shopping requirements efficiently
While exploring different online stores I came across a marketplace that felt clean and minimal with a balanced layout and easy navigation system sun woven collection – Market feels creative with well curated items and smooth navigation, helping users browse products comfortably and quickly.
Many digital learners enjoy platforms that blend inspiration with interaction, especially when browsing curated educational resources, idea boards, and creative development tools frostspire knowledge creation portal – The idea represents a structured learning environment where users can explore new concepts and build creative projects in an engaging and intuitive way.
During my final review of several shopping websites I also found Urban Pick Corner experience hub – The platform was easy to navigate and visually consistent, with a clear structure that helped me move through sections smoothly and understand the layout without any difficulty at all times
During my online browsing time, I stumbled upon browse this page and it felt easy to navigate – The content is well arranged and easy to explore, making it a great browsing experience that feels relaxed and pleasant.
Users exploring digital content platforms usually expect clean structure, focused design, and layouts that make information easy to read and process FocusUltra Flow Node – The UltraFocus platform provides a clear and simple layout, making everything well organized so users can browse content with strong focus and ease.
During my initial browsing session and quick look at different features, I came across this helpful link which gave a good impression – the site looks modern, and the layout feels well structured and visually balanced.
While scrolling through multiple online stores for comparison, I noticed explore trail bazaar – The visit was enjoyable, and everything feels smooth while the information is presented in a clear and easy-to-read way.
While exploring online stores I found something that felt practical and balanced with a modern interface and intuitive navigation structure across categories urban bay browse hub – Goods selection looks practical and well suited for everyday needs, helping users move through pages without confusion.
I wasn’t specifically searching for anything, but I ended up discovering a site that felt worth sharing due to its fresh appearance explore this petal store – The whole site feels fresh, and I like how the products are presented in a simple yet visually pleasing way.
Many online visitors appreciate when they find product choices that stand out during browsing and make them consider coming back to the store at a later time for further review trend center quick view and the experience feels engaging, encouraging users to revisit and continue exploring different categories and items available on the platform.
I was browsing late in the evening when I found a site that felt calm and structured with easy-to-follow category organization silver fern collection hub – I like how everything is categorized, which makes browsing really simple and reduces any confusion while exploring products.
Daily shoppers often look for platforms that update offers regularly while maintaining an easy to use structure that keeps browsing simple and efficient for repeat visits Daily Bargain Space – Here the concept emphasizes a regularly refreshed space where users can find updated bargains in a calm and organized layout designed to make everyday shopping more convenient and enjoyable
Shoppers who prioritize convenience in digital environments usually appreciate platforms that minimize friction while keeping navigation extremely straightforward Shop Portal Access – This service focuses on quick interactions, reducing unnecessary steps and ensuring users can complete purchases without confusion or delays
From my perspective after a short exploration, check it out sits within a well-structured layout – the page feels smooth, and browsing around was pretty enjoyable and easy.
Users exploring online platforms frequently appreciate modern industrial branding, especially when browsing curated essential tools and lifestyle product collections glow forge essentials vision portal – It represents a structured marketplace where products are arranged in a glowing, innovation-inspired environment focused on clarity and purpose.
During my final review of different shopping websites, I focused on overall usability and flow, and Urban Zone Pick site – The platform feels well built, and browsing is comfortable, with a clear layout that makes navigation quick and easy.
Users exploring digital content platforms usually expect clear structure, fast readability, and well organized layouts that simplify browsing and understanding of information LaunchPro Flow Node – The ProLaunch platform appears professional and clean, with content structured nicely for quick reading and easy navigation across all sections of the site.
While exploring online I stumbled upon a store that felt clean and welcoming with a structured interface and smooth browsing system swift maple view hub – Corner shop has cozy feel with surprisingly diverse product range, ensuring a comfortable and easy experience.
While browsing curated lists of learning-focused websites, individuals sometimes highlight that focus guide space appears in resources designed to support easy navigation and clear content presentation for better understanding. – It is often described as user friendly and uncluttered.
Users exploring minimalist online stores often appreciate layouts that emphasize clarity and reduce unnecessary visual complexity Soft Meadow Marketplace allowing them to focus more on products that suit their needs while browsing comfortably and efficiently across a clean and simple interface experience flow
After reviewing a few pages and spending only a short time on the platform, I found that open this site fits into a user-friendly structure – the content appears simple, useful, and accessible to most users.
While reviewing various shopping websites, I discovered explore ember vault – The platform seems solid, and browsing around is straightforward and easy to handle.
As I continued exploring technology-focused platforms, I encountered tech trend insights – a resource that offers daily updates and makes it easier to understand evolving digital and technological changes.
Users exploring online platforms often look for structured interfaces and helpful navigation elements while reviewing digital services through different sources BrandVision portal overview – many users find the BrandVision experience straightforward because its layout emphasizes clarity, and overall browsing becomes more intuitive and efficient for general information access.
While casually browsing different websites I came across something that felt modern and tidy with a balanced layout and clear structure silver field browse collection – Clean design and good layout make this store enjoyable today, making it easy to move through pages without confusion.
I wasn’t expecting to find anything notable, but I came across something that felt simple and well organized while browsing online visit this shop – It’s good for browsing, and the layout is smooth and easy to follow without any confusion or clutter.
People who enjoy unique shopping experiences often prefer platforms that present products in visually appealing and well organized categories for easier discovery Creative Goods Arena – It is portrayed as a creative shopping space where stylish items are displayed in an engaging structure helping users browse efficiently and find interesting products effortlessly
During my exploration of creative websites and knowledge-sharing platforms, I discovered world ideas space – which I enjoy browsing regularly since it always presents something new, interesting, and thought-provoking that keeps the experience refreshing every time.
While checking various digital stores I came across a site that feels modern and organized with clear sections that make it easy to browse different product types urbancrest product lane built with simplicity and usability in mind – The overall experience is smooth, clear, and user friendly
Shoppers often prefer digital stores that provide stable performance and clear navigation paths during product searches and purchases Seamless Cart Space ensuring users can browse comfortably while experiencing fast and reliable system responses throughout their session – The platform prioritizes simplicity and consistency for improved user engagement
While checking out online shops for fun, I discovered visit this page and it felt very organized – The presentation and layout made it simple to find what I needed in a smooth and efficient browsing experience.
People browsing online platforms often look for fast performance, smooth usability, and dynamic interfaces that make exploration simple and enjoyable WaveSharp Access Hub – The SharpWave site feels dynamic and responsive, with navigation working fast and smoothly to provide a seamless browsing experience for users.
During my review of multiple digital shopping interfaces I came across Urban Style Bazaar discover page – The navigation system worked smoothly and everything was clearly arranged, so moving between sections felt natural and the overall experience remained quick, easy, and user friendly throughout the entire visit without complications
During my quick scan of the site and its features, I came across learn more now and appreciated the clarity – the layout looks clean, and the information is easy to follow for users.
While browsing through several online shopping platforms today just to compare layouts and usability, I noticed something in the middle like check this windcrest hub – Everything loads nicely, and the structure feels organized and pleasant overall, making browsing smooth and easy to follow.
While exploring different online stores I came across a platform that felt serene and modern with a balanced interface and neatly organized categories that supported easy browsing twilight cove collection – Collective site gives calm aesthetic and relaxing browsing experience overall, making it feel comfortable and visually gentle.
Users who rely on online tools for daily tasks often prefer platforms that combine stability with straightforward navigation and consistent performance quality TrendPulse Access as usability drives engagement – The system offers a clean and responsive interface that allows users to complete tasks efficiently while maintaining smooth interaction across different sections of the platform
As I browsed through different websites offering development-focused advice, I encountered practical success ideas – a platform that shares grounded growth strategies designed to help people improve step by step without unnecessary complexity.
People searching for handcrafted floral inspired products often value platforms that emphasize artistry, detail, and carefully selected merchandise for home or gifting purposes Artisan Petal Market showcasing a wide array of creative floral themed items designed with attention to detail and quality – the marketplace encourages exploration through its structured layout, intuitive browsing features, and visually appealing presentation that keeps users engaged throughout their visit.
I was casually browsing online when I discovered a store that felt visually distinct with a bold and unusual presentation style silver grove essentials – The interesting concept behind this store stands out from others, giving it a creative and different feel overall.
Modern online buyers increasingly seek digital stores that organize products clearly and reduce browsing effort through structured category layouts and responsive interfaces making shopping faster and more enjoyable across devices and network conditions Center of Style Goods – It serves as a well structured hub where users can explore stylish goods efficiently, benefiting from fast loading pages and an intuitive navigation system designed for convenience
Users exploring branding websites often prefer platforms that feel professional, visually consistent, and easy to navigate across all sections BrandMatrix Experience Link – BrandMatrix delivers strong branding through a modern and polished interface, ensuring a smooth and visually appealing browsing experience for all users.
While researching tools for building online storefronts, I found web commerce builder that offers structured solutions for setting up digital shops – it focuses on flexibility, modular design, and ease of integration, helping developers and business owners create scalable platforms that handle growth efficiently while maintaining clean and manageable architecture for long-term stability.
I wasn’t expecting to find anything notable, but I came across something that felt organized and easy to browse online visit this shop – It’s well structured, and everything is set up to make finding things quick and convenient.
After spending some time exploring different sections and checking usability, I noticed that check this platform fits naturally into the experience – the site looks quite organized, and I found it easy to explore content here overall.
While exploring different e-commerce layouts, I observed that intuitive design matters, and Urban Trend Arena online browsing hub made everything feel straightforward, well structured, and easy to navigate, allowing quick movement through sections without any confusion or delay at all
While researching modern software tools and online knowledge bases, users sometimes mention platforms that focus on clarity and structure like think beyond digital reach which is often referenced as a simple and well organized website. The browsing experience feels consistent and intuitive overall.
While browsing through various websites, I stumbled upon browse copper petal goods and it felt balanced and neat – Browsing here was enjoyable, and the items seem interesting and worth checking again due to the selection.
While checking different online stores I found a site that feels modern and structured with a visually appealing layout that keeps browsing simple and efficient for users urban harbor goods hub featuring clean category organization – Products are displayed in a visually pleasing and easy to understand way
During my quick review of the platform and its various sections, I came across learn more now and appreciated the clarity – the experience feels smooth, and the accessibility makes navigation simple.
I had been exploring different sites when I found open wind petal market – The interface is clean and simple, and it improves the browsing experience noticeably throughout.
Marketing agencies often rely on integrated platforms that help manage campaigns, track analytics, and improve overall digital performance Growth Optimization Suite allowing teams to refine strategies, boost engagement, and achieve more predictable results across multiple client projects consistently over time
Many online audiences expect websites to provide both fast loading speed and a structured interface that improves clarity and overall navigation experience UltraConnect UX Node – UltraConnect features a modern design where usability and speed work together, ensuring users can navigate smoothly and access content without difficulty.
Those who actively seek personal growth often find value in exploring structured advice that promotes healthier routines, and a helpful example can be found at everyday-wellness-notes – The material there encourages small but meaningful adjustments that gradually enhance mental clarity, productivity, and overall satisfaction with daily living experiences.
I was navigating through different websites when I found shop collection now and it looked clean and organized – I enjoyed visiting this site because the items appear trendy and worth checking out, creating a smooth browsing experience.
During my time exploring industry news websites, I encountered trend news center – which provides valuable insights and updates that help me stay informed about evolving market conditions and developments.
Users exploring websites typically appreciate platforms that feel safe, well designed, and easy to navigate without unnecessary complexity or visual clutter BrightVault Navigation Node – The BrightVault platform provides a secure and clean interface where users can browse comfortably while enjoying a visually appealing and structured layout.
I was casually browsing online when I found a store that felt visually balanced with a soft and simple layout design approach silver harbor essentials – Smooth browsing experience and products seem thoughtfully selected here, creating a comfortable and easy shopping flow.
While casually searching the internet I discovered a site that felt modern and refined with a balanced layout and clearly organized product sections twilight crest goods store – Store feels polished and easy to navigate across all sections, helping users find items quickly.
Many online visitors appreciate websites that group products neatly and allow fast switching between categories for better shopping efficiency Simple Style Hub this platform highlights simple structure and smooth performance making it easier for users to locate stylish items without wasting time during browsing sessions
People browsing e-commerce platforms usually appreciate systems that reduce search time and highlight relevant promotions based on trending demand and popularity Speedy Deals Access – The platform enhances efficiency by offering quick navigation tools and a responsive layout that allows shoppers to move directly to the most valuable discounts available at any moment.
I wasn’t specifically searching for anything, but I ended up finding something that felt worth sharing after browsing through it for a while explore this thread site – The shop seems nice, and the items look unique with a thoughtful and creative approach behind them.
Many customers searching for handmade lifestyle products tend to appreciate digital stores that emphasize wood textures and earthy tones in their collections Pinewood Craft Collective offering thoughtfully arranged rustic items and creative home accessories that reflect traditional artistry – the overall experience feels grounded and warm, with a strong emphasis on simplicity and natural craftsmanship.
While casually browsing and testing different features, I noticed that follow this link contributes to a smooth experience – the design feels user friendly, and everything is organized in a clear and accessible way.
In between browsing different platforms, I noticed visit windspire collective now – The platform feels reliable overall, and the content is smooth and easy to navigate from start to finish.
Users exploring web platforms often prefer systems that reduce instability and ensure smooth access to content through reliable and well structured interfaces WebStable Experience Portal – It offers a clean layout with stable performance and intuitive navigation that helps users browse content efficiently while maintaining a comfortable and uninterrupted browsing experience across all pages
While exploring different online stores I discovered a website that feels modern and clean with a themed design that enhances product visibility and browsing ease lighthouse goods urban hub featuring simple navigation – The store has a unique concept with neatly arranged product listings
Users interacting with digital platforms typically value simplicity, organization, and premium design that enhances readability and user flow EliteZone Navigation Node – EliteZone provides a well structured and premium interface where browsing feels easy, organized, and visually clean throughout all pages.
I was casually browsing online when I found a site that felt responsive and clean with a layout that made everything easy to access quickly silver horizon essentials – Everything loads quickly and interface feels very user friendly today, creating a comfortable and efficient browsing experience.
Modern shoppers frequently look for platforms that present goods clearly while maintaining responsive performance across different devices and browsing conditions Modern Goods Point – This point structure ensures clarity in product presentation and supports a smooth transition from browsing to checkout completion
As I searched for intuitive and easy-to-use platforms, I came across smooth interface guide – which offers structured information and a minimal design that enhances readability and creates a seamless browsing experience.
While researching modern online marketplaces and shopping interfaces, I discovered smart tree shopping guide that helps users browse products through clear categories – Shop tree market feels organized with useful categories for shoppers and improves overall shopping flow by offering a clean structure that reduces confusion and supports faster decision-making processes.
In discussions about user centered browsing platforms and simplified digital layouts, users occasionally refer to idea orbit panel as part of collections that emphasize clarity, structure, and smooth navigation for general online content exploration. – The design is clean, steady, and easy to follow.
While reviewing the interface and moving through sections, I came across go to this site which fits into a neat structure – the layout is interesting, and everything feels simple and easy to navigate around.
While testing various digital shopping experiences I focused on speed and interface design and Urban Trend Hub online shop – Everything seems well structured and dependable, with pages loading smoothly and a clean design that supports comfortable browsing without unnecessary visual clutter
As I moved through pages and explored features, I found that view more info integrates into a tidy structure – the first impression is strong, and the layout feels modern and approachable for users.
During random browsing I stumbled upon a shop that felt green inspired and tidy with a clean interface and simple navigation flow twilight fern shop hub – Fern themed store looks natural fresh and visually pleasing overall, making browsing efficient and easy.
While exploring various websites today, I discovered something that felt calm and visually soft enough to share in this discussion browse this emporium – The vibe is peaceful, and the products appear curated in a way that makes the whole experience feel smooth and relaxing.
In the middle of exploring idea-based learning platforms, I discovered simple answers guide – a site that focuses on making information easy to understand while helping users explore useful ideas and quick solutions.
While going through different craft websites, I encountered check azure items and it felt calm and structured – The crafts selection feels creative and well organized, giving the browsing experience a relaxed and pleasant feel.
During a casual search session, I found myself checking out open this page – It gives a clean impression, with a straightforward design that allows users to move through the content comfortably and without any confusion.
During my review of brand strategy tools I focused on usability and system design and found BuildYourBrand creative engine – The platform provides useful and easy to understand tools, making branding work efficient and straightforward while ensuring a smooth and intuitive user experience overall
Shoppers drawn to seaside collectibles often enjoy browsing curated stores that focus on natural coastal themes for global online buyers today Shoppers drawn to seaside collectibles often enjoy browsing curated stores that focus on natural coastal themes for global online buyers today Seashell Curated Shop featuring elegant shell inspired decor and handmade items – A curated coastal shop designed for collectors and decor enthusiasts today
Digital visitors often prefer websites that feel organized and visually pleasant, allowing them to focus on content while navigating easily through pages NexusUrban Navigation Hub – The UrbanNexus platform offers a clean and structured layout with engaging content, ensuring users enjoy a smooth and visually appealing browsing journey overall.
Users generally prefer platforms that maintain consistency in design and offer straightforward navigation experiences < ImpactDrive Alpha – Alphaimpact ensures a strong interface design where information is clearly structured, allowing users to browse easily and efficiently with a smooth user experience overall.
While casually browsing different websites I came across something that felt modern and minimal with a well structured and clear layout silver lane browse collection – I found this shop quite easy to use and navigate around, making it easy to move through pages without confusion.
Modern consumers often look for platforms that support seamless browsing across devices with responsive layouts and organized content Responsive Style Zone – This version highlights adaptability and ease of use ensuring users can browse stylish goods smoothly whether on mobile or desktop
Shoppers who prefer premium online experiences often look for stores that combine elegance with straightforward navigation and thoughtfully categorized product listings Premium Lifestyle Outlet – the website presents a refined collection of household goods that blend modern aesthetics with practical everyday usability in a seamless browsing flow
In articles discussing boutique marketplace innovations, readers frequently encounter contextual links integrated within narrative explanations of brand identity, especially when they come across urban petals collective storefront guide – Such storefront guides are known for helping users understand product categories through visually consistent layouts and structured browsing paths that support discovery without overwhelming complexity.
During my analysis of shopping site usability I evaluated layout clarity and navigation flow and UTS online shop entry – I enjoyed browsing here, structure makes it easy to look around, and everything felt arranged logically so finding items or sections did not require extra effort or repeated searching.
During my browsing of educational and informational sites, I discovered simple learning grid – which offers clearly structured content that supports easy understanding and smooth learning progression for all types of users.
After reviewing multiple sections and paying attention to the layout, I found that open this site fits within a well-organized structure – the interface feels clean, making it easy to find things without delay.
During a casual browsing session where I was comparing a few platforms, I ended up encountering explore this shop – The way everything is organized seems intuitive and clean, making it easy to move through pages without feeling overwhelmed at any point.
oakridgeemporium.shop – This store caught my attention, design is simple yet appealing.
Writers designers and innovators often prefer digital environments that allow seamless collaboration and idea exchange and a notable example is Creative Flow Network which enhances productivity and creative synergy – A connected digital ecosystem designed to improve workflow and encourage continuous sharing of innovative ideas
People evaluating websites often focus on innovation, simplicity, and how effectively the interface communicates information through a clean structure LabsZenith Interface Hub – The ZenithLabs platform looks innovative with a simple yet effective layout, making it easy for users to understand and navigate content.
While casually searching the internet I discovered a site that felt clean and modern with an intuitive layout and easy category flow silver oak goods store – Overall good experience browsing through items and checking different categories, giving a simple and efficient browsing experience.
As I navigated through different sections and usability flow, I noticed that visit here aligns with a structured interface – good site overall, with easy navigation and pages loading without issues.
Online shoppers often look for platforms that prioritize selected items and reduce unnecessary browsing steps for a more efficient shopping experience Quick Style Picks – This approach ensures that curated products are displayed clearly with fast navigation tools that help users locate items without wasting time
Users who prefer clean and modern beach inspired shopping environments often seek platforms that deliver both simplicity and usefulness in their offerings Beachside Minimal Store presenting carefully selected products that reflect coastal living and minimalist design principles – the marketplace ensures an easy shopping journey with uncluttered layout, functional items, and a calm aesthetic suitable for everyday needs
Students of self-development frequently look for digital spaces that combine clarity with motivation, especially when they discover resources like motivation-boost-center – that focus on pushing individuals beyond their comfort zones while offering supportive guidance, helping them maintain momentum and develop stronger discipline in everyday routines.
I randomly clicked through a few pages and landed on explore this store which seemed nicely designed – It’s a really pleasant browsing experience where everything is neat, clean, and thoughtfully arranged in a visually easy way.
Online consumers increasingly expect platforms that load quickly and allow seamless movement between categories while maintaining a clean and user friendly interface Fast Checkout Lane – It delivers a simplified checkout system that reduces waiting times and helps users complete transactions efficiently without unnecessary interruptions or complicated navigation paths
During my browsing session and usability check, I noticed that see more here blends into a clear layout – the platform feels nice, and content appears relevant and well presented overall.
While browsing the site and reviewing how information is presented across pages, I found that visit this resource stands out in a subtle way – everything feels fresh, and the content is arranged in a clear and relevant manner that is easy to follow.
While reviewing various online shopping platforms for usability I noticed a clean layout and smooth navigation that stood out when visiting Urban Trend Zone official page making browsing feel easy and intuitive across categories – Good experience overall, design is simple and quite user friendly with clear structure and minimal distractions throughout the visit
Users who value efficiency in online shopping tend to prefer stores that keep things minimal and direct, such as practical shopping navigation site – The browsing experience is intentionally simple, allowing customers to explore available items with ease while the structure remains clean and focused on functional usability rather than decorative complexity.
While scrolling through multiple shopping pages out of curiosity, I noticed open this site – The layout appears clean and responsive, and the pages load without noticeable issues, making it easy to explore content at a steady pace.
For those interested in enhancing their creative thinking and branding skills, encountering see branding insights can be a great opportunity – delivering inspiration along with practical, easy-to-follow examples that support deeper understanding.
People evaluating online systems often focus on how creatively ideas are presented and whether the interface supports simple and intuitive navigation GeniusNew Clean Portal – The NewGenius platform feels creative and easy to use, making exploration simple, structured, and comfortable for users across all pages.
Frontend developers often seek optimized systems that reduce latency and improve the responsiveness of interactive elements within applications FastTrack Media Studio commonly discussed in development forums – It provides a smooth operational structure that enhances performance and allows quicker content delivery across various platforms
I came across something while exploring online that felt straightforward and worth mentioning, so I decided to share it here check this site – It has a decent selection overall, and everything seems easy to browse and understand without any complexity.
Many digital users expect websites to maintain a balance between simplicity, speed, and clear organization for a better overall experience PowerCore Interface Link – The PowerCore platform offers a strong design approach where navigation feels intuitive, structured, and efficient across all areas of the site.
During my usual browsing session I came across a site that felt warm and rustic with a structured interface and clearly organized categories twilight grove market – Goods here feel thoughtfully chosen with appealing rustic style vibe, offering a calm and easy browsing flow.
At some point during my online reading, I encountered business idea expansion embedded in the article – It helped me see improvement strategies in a more structured and practical way than before.
Modern consumers tend to value platforms that keep them updated with current trends while offering a seamless browsing experience across devices Current Style Hub – The hub focuses on showcasing the latest product ideas with a responsive and organized interface designed for convenience
After analyzing how content is structured and delivered, I found that see this website fits into a well-arranged layout – browsing felt good, and everything loaded smoothly with clear visual presentation.
While analyzing e-commerce platforms aimed at cost-conscious users, I encountered low price market hub that simplifies product discovery through structured layouts – Value market hub focuses on affordable items and easy navigation, helping users access budget-friendly options quickly while maintaining a smooth and organized shopping experience across multiple product sections.
During my review of digital storefronts I noticed a strong emphasis on clarity in some platforms including ValueFactory product hub – The interface maintains a clean look and easy flow, making the site feel pleasant to use while supporting straightforward navigation across all sections without difficulty
I was exploring different websites for comparison when I found enter this page – The platform seems responsive, and its structured layout makes navigation simple and clear.
Shoppers exploring environmentally conscious platforms often value stores that highlight ethical production and plant based inspiration in product design Sprout Harmony Eco Store offering curated sustainable items that blend functionality with eco friendly materials and practices – It provides a user focused shopping environment designed for greener and healthier lifestyle choices
People reviewing online systems usually prioritize how well ideas are linked together, how clean the interface appears, and whether navigation feels intuitive and simple BridgeMax Interface Node – The Maxbridge platform connects ideas in a smooth way with a clean design, ensuring users can navigate easily and access information without unnecessary complexity.
Digital motivation platforms are valuable for individuals who want to stay consistent in their goals while receiving encouragement that reinforces positive behavioral change Consistency Growth Portal plays this role – It supports users in maintaining steady progress and building confidence through structured motivational content and goal oriented reinforcement strategies
In the process of discovering valuable online spaces, users may appreciate finding visit this destination – a platform that delivers engaging content alongside a smooth and intuitive browsing experience that makes every visit enjoyable and worthwhile.
I was navigating through different websites when I found shop collection now and it looked clean and structured – I found some cool items here and I’ll definitely be returning again to browse further in the future.
While exploring online networking spaces, I found people connect space – which offers a platform where users can interact, share experiences, and learn from each other in a supportive environment.
While reading different creative discussions online I discovered idea creation hub within the content and it immediately drew my attention – It offered an interesting perspective that felt worth exploring further because of its practical and thoughtful approach.
During my quick scan of the website and structure, I came across learn more now and appreciated the layout – the design looks professional, and everything is neatly arranged and clean.
лечение алкоголизма вызов на дом [url=https://narkolog-na-dom-samara-9.ru]https://narkolog-na-dom-samara-9.ru[/url]
People comparing ecommerce websites usually focus on speed and organization as key factors, and in such evaluations they may discover pine goods digital catalog hub – Products appear well structured into clear categories, and the site responds quickly, creating a browsing experience that feels efficient, lightweight, and easy to use across all sections of the platform.
Many users browsing online stores prefer environments that highlight current styles while maintaining a clutter free design for smoother navigation across product categories Trend Style Corner Spot – The experience centers around displaying trendy goods in an organized layout that supports quick browsing and comfortable user interaction
While casually browsing and testing features, I noticed that follow this link contributes to a smooth experience – the design looks appealing, and navigation feels simple, fast, and highly intuitive throughout.
While exploring digital retail systems focused on convenience, I found a browsing enhancement platform quick product portal that organizes listings clearly and improves user navigation across various shopping categories – Quick online market supports efficient browsing and provides users with a smoother experience when searching for relevant products online without unnecessary complexity.
капельница на дому в екатеринбурге [url=https://kapelnicza-ot-pokhmelya-ekaterinburg-14.ru]https://kapelnicza-ot-pokhmelya-ekaterinburg-14.ru[/url]
While reviewing layout and testing navigation, I saw that open this link integrates into a clear system – a quick look suggests the design is clean, modern, and smooth in appearance today.
During my exploration of e-commerce platforms I focused on usability flow and layout clarity and found Value Goods Bazaar browse portal – The interface feels neat and organized, and browsing through was actually pretty smooth, with clear structure that helps users find content quickly without unnecessary complexity or confusion
While exploring online shopping platforms, I discovered check this page – The experience is smooth, with a clean design and easy-to-follow navigation.
Users comparing online platforms often prioritize responsiveness, visual consistency, and how effectively the design adapts across different screen sizes and browsing contexts EliteFusion Navigation Portal – EliteFusion delivers a modern and responsive interface where design is blended beautifully, making browsing smooth, visually attractive, and easy to understand for all users.
Readers interested in productivity often appreciate reminders that highlight the importance of valuing each moment in a practical way value each moment post the message feels encouraging and easy to understand, making it relatable – it reinforces the idea that consistent awareness leads to more meaningful daily experiences
During random browsing I stumbled upon a shop that felt tidy and cozy with a structured interface and simple navigation flow twilight maple shop hub – Corner shop gives warm atmosphere and easy product discovery experience, making browsing efficient and straightforward.
капельницы на дому екатеринбург цена [url=https://kapelnicza-ot-pokhmelya-ekaterinburg-16.ru]капельницы на дому екатеринбург цена[/url]
People working on performance improvement often rely on structured systems that help them organize tasks and maintain focus on outcomes AchieveEdge Hub – A results driven platform that encourages users to refine their workflow habits while staying committed to their goals and improving productivity through consistent action oriented planning and execution support tools
While analyzing web performance tools and interface efficiency examples, people frequently refer to ascendmark clean access which appears in collections highlighting minimal design and fast response behavior across modern browsing environments. – The layout is typically seen as smooth, quick, and very stable.
As I browsed through different innovation-focused websites, I encountered future insight space – a platform that presents content with a forward-looking approach, helping digital users stay informed about emerging concepts and evolving online landscapes in a meaningful way.
I was casually scrolling through educational content when I found easy success space placed in the middle of the page and it stood out – The content feels clean and simple, which makes it very easy to follow and absorb without effort.
People searching for rustic home solutions often value marketplaces that combine earthy aesthetics with durable construction and long term usability for daily needs SpruceField Lifestyle Market featuring practical goods influenced by forest environments – The store ensures an engaging shopping experience centered on natural design principles and functional products
Consumers exploring seasonal styles usually look for platforms that gather trending items into one place with clear organization and easy navigation Seasonal Trend Portal – This portal organizes seasonal updates into accessible categories, improving browsing efficiency
After examining the page layout and reading through the available material, I observed that this useful link fits within a clean design – the text is clear, and the structure enhances overall readability.
Погружайся в захватывающие сюжеты вместе с нами! Голливудские блокбастеры, культовые сериалы, добрые мультфильмы и зрелищные премьеры – всё доступно в отличном качестве. Никакой рекламы, только чистое удовольствие от просмотра. Создай свою коллекцию любимых фильмов и наслаждайся: сериалы онлайн
People browsing for online shopping solutions appreciate platforms that reduce complexity and improve overall clarity in product presentation NextGen Market Place – The website is designed to support effortless navigation, ensuring users can quickly find items and enjoy a smooth shopping experience
While testing different online retail websites I observed navigation quality and layout structure and found VCC storefront page – Nice little setup, everything looks clear and simple to understand, with a straightforward interface that allows users to browse comfortably without unnecessary distractions or confusion
People evaluating digital platforms often focus on visual harmony, usability, and how well design adapts to different screens and navigation patterns FusionElite Interface Hub – The EliteFusion website combines design beautifully, offering a modern and responsive experience where users can navigate easily and enjoy a clean visual flow.
While going through a variety of online shops, I encountered check haven items and it felt calm and organized – The marketplace feels friendly, easy to navigate, and quite enjoyable overall, making browsing feel effortless and pleasant.
While looking for informative resources about opportunities, I came across growth discovery guide – a platform that shares insights on where and how to find valuable opportunities that can lead to personal and professional advancement.
People exploring curated ecommerce sites frequently appreciate when design and usability work together seamlessly, particularly when they encounter soft elegance shopping portal – The store feels elegant with its soft aesthetic styling and provides easy navigation, making it simple for users to browse products comfortably and enjoy a smooth and visually pleasant experience throughout the platform.
Organizations worldwide depend on efficient communication tools that unify workflows and improve coordination between remote teams and global operations daily usage flow Integrated Global Access Portal supporting seamless digital interaction and fast performance globally efficiently now – It delivers structured navigation that enhances global system usability and user experience efficient smooth worldwide access
Digital strategists working on scaling presence often look for advanced approaches, and in that development stage Presence Scaling Strategy handbook – it is generally described as a structured resource that helps optimize growth processes, improve visibility systems, and expand reach efficiently over time.
In various usability discussions and interface evaluations, people sometimes refer to lightweight platforms designed for efficiency, including sharp bridge overview hub which is listed among tools focused on simple browsing and clear content accessibility. – The experience is clean and very straightforward.
While exploring online stores I found a site that felt earthy and minimal with a clean layout and easy category navigation system twilight meadow collection view – Goods feel nature inspired and browsing experience remains very pleasant, giving a relaxed and smooth browsing experience overall.
As I searched for well-explained media resources, I came across media learning hub – which offers helpful insights presented in a simple way, allowing users to easily understand important topics in modern media.
As I navigated through different sections and observed usability, I noticed that visit here aligns with a structured interface – I enjoyed the visit, and the layout and organization feel quite decent and straightforward.
While going through random posts online earlier today, I unexpectedly came across this cool story hub and the experience was surprisingly pleasant and immersive – It felt like each piece was written to keep curiosity alive and maintain a smooth reading flow throughout.
Users interested in efficient shopping experiences typically prefer platforms that minimize waiting time and offer clear product listings across all sections Instant Goods Corner – The corner emphasizes rapid loading and easy navigation, ensuring users can browse items without interruptions
People reviewing online systems usually prioritize clarity of presentation, functional design, and how easily they can understand and navigate content without unnecessary complexity VisionMax Interface Node – The MaxVision platform provides a clear visual experience where design is simple and highly functional, ensuring users can browse smoothly and understand information quickly.
People exploring online fashion stores usually expect a smooth interface that highlights trending outfits and seasonal discounts in a structured format Style Deal Navigator this platform enhances user convenience by offering fast loading pages and simplified navigation paths for discovering stylish wardrobe options easily
As I browsed casually through different sections, I saw that click for details aligns with a tidy structure – this site looks promising, and the layout and usability feel decent overall.
During my analysis of online retail websites I focused on structure and navigation flow and found Vivid Cart Corner digital portal – Overall a good visit, layout feels modern and easy to navigate, providing a comfortable browsing experience where users can move between pages efficiently without confusion
People who appreciate raw industrial charm often browse curated shops that introduce floral inspired artistry into metal based home accessories Rust & Bloom Industrial Corner offering carefully selected pieces that combine steel aesthetics with organic inspiration – The store highlights a unique design philosophy that merges toughness with natural softness in everyday décor
Many people interested in positivity and self-improvement turn to inspire and grow space – a helpful resource that shares uplifting content and practical ideas to support consistent personal development and a stronger mindset.
People aiming to grow their online ventures often rely on platforms that offer simplified explanations and step by step guidance for sustainable improvement Growth Insight Studio practical knowledge sharing theme – supporting users with clear direction and useful strategies that help them achieve better outcomes and long term business success efficiently.
For users who enjoy educational content, visiting learning share guide – a platform that provides fresh and engaging material can help individuals stay motivated while exploring new topics and exchanging ideas with a wider community.
People interested in self development often find that simple motivational cues help them overcome procrastination effectively growth momentum key – this idea emphasizes that consistent action is more powerful than occasional effort and helps build strong long term habits naturally over
Users exploring online systems typically expect smooth navigation and logically arranged information that helps them find what they need quickly and efficiently NovaStream Interface Hub – The NovaOrbit design focuses on futuristic presentation and well-organized content flow, making exploration enjoyable and reducing friction during browsing sessions.
People comparing online stores frequently look for clarity and ease of navigation, particularly when they discover crest product browsing portal – The market maintains a clean interface with good product arrangement and clear presentation, helping users enjoy a smooth and simple shopping experience without unnecessary distractions or complicated interface elements.
I had some free time and decided to explore a few sites when I discovered discover haven store and it looked very clean – The simple design and clear listings made browsing here smooth, convenient, and straightforward in a very natural way.
As I examined the interface and content flow, I noticed that this page here contributes to a simple experience – the content appears interesting, updated, and relevant for users today overall.
Users who prefer fast and straightforward shopping experiences often look for platforms that reduce delays and improve navigation flow across categories Straightforward Goods Zone – The platform provides easy browsing with efficient product access and minimal effort
Many digital audiences appreciate platforms that combine strong visual design with clean layouts that enhance readability and user engagement VisionMark Experience Link – VisionMark delivers strong visuals with a clean and well structured interface, ensuring users can explore content comfortably and understand information quickly.
Users who prefer optimized online systems often rely on efficient retail selection tool – Cart choice store is built to improve speed and accuracy in shopping decisions, helping customers quickly identify and choose products that best fit their needs and preferences
If you’re searching for a place to stay motivated and learn from others, checking out grow and inspire hub – a resource offering uplifting insights and practical encouragement can help maintain steady progress in everyday life goals.
Shoppers looking for creative industrial décor often prefer marketplaces that highlight fusion of steel structures and delicate botanical themes in products SteelBloom Interior Craft Hub showcasing distinctive handmade items designed for stylish living spaces – The platform emphasizes artistic contrast and innovative design thinking for home improvement inspiration
Online shoppers often appreciate websites that present curated selections in a logical layout to improve browsing speed and overall experience Focused Picks Corner – This corner is designed to highlight key items and ensure easy access through simple navigation
Shoppers who want a simple online experience typically prefer platforms that keep product selection straightforward and checkout processes efficient Simple Choice Market – The design emphasizes ease of use and fast transactions
While analyzing various platforms for online store management, I encountered ecommerce zone builder which offers structured solutions for digital retailers – it supports improved store customization, easier navigation systems, and flexible tools that help businesses manage their online operations while maintaining scalability and smooth performance across different store setups.